Unveiling Human Non-Random Genome Editing Mechanisms Activated in Response to Chronic Environmental Changes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Activation-Induced Deoxycytidine Deaminase (AID) Co-Transcriptional Scanning at Single-Molecule Resolution
ARTICLE Received 19 Nov 2014 | Accepted 13 Nov 2015 | Published 18 Dec 2015 DOI: 10.1038/ncomms10209 OPEN Activation-induced deoxycytidine deaminase (AID) co-transcriptional scanning at single-molecule resolution Gayan Senavirathne1,*, Jeffrey G. Bertram2,*, Malgorzata Jaszczur2,*, Kathy R. Chaurasiya3,4,*, Phuong Pham2, Chi H. Mak5,6, Myron F. Goodman2,5 & David Rueda1,3,4 Activation-induced deoxycytidine deaminase (AID) generates antibody diversity in B cells by initiating somatic hypermutation (SHM) and class-switch recombination (CSR) during transcription of immunoglobulin variable (IgV) and switch region (IgS) DNA. Using single- molecule FRET, we show that AID binds to transcribed dsDNA and translocates unidirectionally in concert with RNA polymerase (RNAP) on moving transcription bubbles, while increasing the fraction of stalled bubbles. AID scans randomly when constrained in an 8 nt model bubble. When unconstrained on single-stranded (ss) DNA, AID moves in random bidirectional short slides/hops over the entire molecule while remaining bound for B5 min. Our analysis distinguishes dynamic scanning from static ssDNA creasing. That AID alone can track along with RNAP during transcription and scan within stalled transcription bubbles suggests a mechanism by which AID can initiate SHM and CSR when properly regulated, yet when unregulated can access non-Ig genes and cause cancer. 1 Department of Chemistry, Wayne State University, 5101 Cass Avenue, Detroit, Michigan 48202, USA. 2 Department of Biological Sciences, University of Southern California, Los Angeles, California 90089, USA. 3 Department of Medicine, Section of Virology, Imperial College London, Du Cane Road, London W12 0NN, UK. 4 Single Molecule Imaging Group, MRC Clinical Sciences Center, Imperial College London, Du Cane Road, London W12 0NN, UK. -

Difluorodeoxycytidine 5'-Triphosphate: a Mechanism of Self-Potentiation1
ICANCER RESEARCH 52, 533-539, February 1, 1992] Cellular Elimination of 2',2'-Difluorodeoxycytidine 5'-Triphosphate: A Mechanism of Self-Potentiation1 Volker Heinemann,2 Y¡-ZhengXu, Sherri Chubb, Alina Sen, Larry W. Hertel, Gerald B. Grindey, and William Plunkett1 Department of Medical Oncology, The University of Texas M. D. Anderson Cancer Center, Houston, Texas 77030 [V. H., Y. X., S. C., A. S., W. P.], and Lilly Research Laboratories, Indianapolis, Indiana 46285 (L. W. H., G. B. G.] ABSTRACT less, clinical cellular pharmacology studies have demonstrated 2',2'-Difluorodeoxycytidine (dFdC, Gemcitabine) is a deoxycytidine that the dFdCTP:dCTP value reaches potentially inhibitory analogue which, after phosphorylation to the 5'-di- and 5'-triphosphate values during clinical trials (4, 10, 11). (b) dFdCTP is incor porated into DNA by DNA polymerases a and f, inhibiting (dFdCTP), induces inhibition of DNA synthesis and cell death. We examined the values for elimination kinetics of cellular dFdCTP and further elongation (9). (c) Once incorporated, dFdCMP resi found they were dependent on cellular concentration after incubation of dues in the terminal or penultimate positions of the DNA strand CCRF-CEM cells with dFdC and washing into drug-free medium. When inhibit the editing function of DNA polymerase <(9). This may the drug was washed out at low cellular dFdCTP levels (<50 n\\), fix damage caused by the incorporated analogue, (d) dFdCDP dFdCTP elimination was linear (t,: = 3.3 h), but it became biphasic at inhibits ribonucleotide reducÃase,blocking DNA synthesis by intracellular dFdCTP levels >100 JIM. Although the initial elimination decreasing the cellular concentrations of deoxynucleoside tri rate was similar at all concentrations, at higher concentrations the phosphates (12-14). -

Determining HDAC8 Substrate Specificity by Noah Ariel Wolfson A
Determining HDAC8 substrate specificity by Noah Ariel Wolfson A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biological Chemistry) in the University of Michigan 2014 Doctoral Committee: Professor Carol A. Fierke, Chair Professor Robert S. Fuller Professor Anna K. Mapp Associate Professor Patrick J. O’Brien Associate Professor Raymond C. Trievel Dedication My thesis is dedicated to all my family, mentors, and friends who made getting to this point possible. ii Table of Contents Dedication ....................................................................................................................................... ii List of Figures .............................................................................................................................. viii List of Tables .................................................................................................................................. x List of Appendices ......................................................................................................................... xi Abstract ......................................................................................................................................... xii Chapter 1 HDAC8 substrates: Histones and beyond ...................................................................... 1 Overview ..................................................................................................................................... 1 HDAC introduction -

B Number Gene Name Mrna Intensity Mrna Present # of Tryptic
list list sample) short list predicted B number Gene name assignment mRNA present mRNA intensity Gene description Protein detected - Membrane protein detected (total list) detected (long list) membrane sample Proteins detected - detected (short list) # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides # of tryptic peptides Functional category detected (membrane Protein detected - total Protein detected - long b0003 thrB 6781 P 9 P 3 3 P 3 0 homoserine kinase Metabolism of small molecules b0004 thrC 15039 P 18 P 10 P 11 P 10 0 threonine synthase Metabolism of small molecules b0008 talB 20561 P 20 P 13 P 16 P 13 0 transaldolase B Metabolism of small molecules b0009 mog 1296 P 7 0 0 0 0 required for the efficient incorporation of molybdate into molybdoproteins Metabolism of small molecules b0014 dnaK 13283 P 32 P 23 P 24 P 23 0 chaperone Hsp70; DNA biosynthesis; autoregulated heat shock proteins Cell processes b0031 dapB 2348 P 16 P 3 3 P 3 0 dihydrodipicolinate reductase Metabolism of small molecules b0032 carA 9312 P 14 P 8 P 8 P 8 0 carbamoyl-phosphate synthetase, glutamine (small) subunit Metabolism of small molecules b0048 folA 1588 P 7 P 1 2 P 1 0 dihydrofolate reductase type I; trimethoprim resistance Metabolism of small molecules peptidyl-prolyl cis-trans isomerase (PPIase), involved in maturation of outer b0053 surA 3825 P 19 P 4 P 5 P 4 P(m) 1 GenProt membrane proteins (1st module) Cell processes b0054 imp 2737 P 42 P 5 0 0 P(m) 5 GenProt organic solvent tolerance Cell processes b0071 leuD 4770 -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Induction of Deoxycytidine Deaminase Activity in Mammalian Cell Lines By
Proc. Nati. Acad. Sci. USA Vol. 74, No. 4, pp. 1734-1738, April 1977 Microbiology Induction of deoxycytidine deaminase activity in mammalian cell lines by infection with herpes simplex virus type 1 (mouse mutant cell lines/selective system/thymidine kinase/antiviral chemotherapy) TEH-SHENG CHAN Department of Physiology, University of Connecticut Health Center, Farmington, Connecticut 06032 Communicated by E. A. Adelberg, January 26,1977 ABSTRACT Herpes simplex virus type 1 induces deoxycy- activity when it lytically infects dCD- mouse cells. It will also tidine deaminase (cytidine/deoxycytidine aminohydrolase, EC be shown that the induced enzyme is very likely coded by the 3.5.4.5) activity when it lytically infects a number of mammalian is a useful cell lines. The deaminase activity is induced in a mouse cell line viral genome. We have previously shown that dCD that is deficient in this enzyme. The induction of the enzyme selective marker for somatic cell hybridization (14). Thus, this in this mutant cell line does not occur in the presence of acti- enzyme can also be used as a selective marker for isolating nomycin D and the induced enzyme is more thermolabile than dCD+ cells from dCD- cells after infection by UV-inactivated the enzyme.of the wild-type mouse cell line. Furthermore, a new HSV-1 virus. deoxycytidine deaminase species with a characteristic elec- trophoretic mobility that is different from that of the host cell enzyme is found in cell extracts prepard from a human cell line MATERIALS AND METHODS infected with herpesvirus. These results strongly suggest that the virus-induced deoxycytidine deaminase is coded by the viral Cells. -

The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors After Cerebral Ischemia: Facts and Hypotheses
International Journal of Molecular Sciences Review The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses Svetlana Demyanenko 1,* and Svetlana Sharifulina 1,2 1 Laboratory of Molecular Neurobiology, Academy of Biology and Biotechnology, Southern Federal University, pr. Stachki 194/1, 344090 Rostov-on-Don, Russia; [email protected] 2 Neuroscience Center HiLife, University of Helsinki, Haartmaninkatu 8, P.O. Box 63, 00014 Helsinki, Finland * Correspondence: [email protected]; Tel.: +7-918-5092185; Fax: +7-863-2230837 Abstract: Histone deacetylase (HDAC) and histone acetyltransferase (HAT) regulate transcription and the most important functions of cells by acetylating/deacetylating histones and non-histone proteins. These proteins are involved in cell survival and death, replication, DNA repair, the cell cycle, and cell responses to stress and aging. HDAC/HAT balance in cells affects gene expression and cell signaling. There are very few studies on the effects of stroke on non-histone protein acetylation/deacetylation in brain cells. HDAC inhibitors have been shown to be effective in protecting the brain from ischemic damage. However, the role of different HDAC isoforms in the survival and death of brain cells after stroke is still controversial. HAT/HDAC activity depends on the acetylation site and the acetylation/deacetylation of the main proteins (c-Myc, E2F1, p53, Citation: Demyanenko, S.; ERK1/2, Akt) considered in this review, that are involved in the regulation of cell fate decisions. Sharifulina, S. The Role of Post-Translational Acetylation and Our review aims to analyze the possible role of the acetylation/deacetylation of transcription factors Deacetylation of Signaling Proteins and signaling proteins involved in the regulation of survival and death in cerebral ischemia. -

Alzheimer's Disease As a Chronic Maladaptive Polyamine Stress
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 17 February 2021 doi:10.20944/preprints202102.0362.v1 Alzheimer’s disease as a chronic maladaptive polyamine stress response Review Baruh Polis1, David Karasik2,3, Abraham O. Samson1 1 Drug Discovery Laboratory, The Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel. 2 Hebrew SeniorLife, Hinda and Arthur Marcus Institute for Aging Research, Boston, MA, 02131, USA 3 Musculoskeletal Genetics Laboratory, The Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel. Correspondence to Baruh Polis [email protected] Keywords: polyamines; arginase; senescence; aging; neurodegeneration. 1 © 2021 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 17 February 2021 doi:10.20944/preprints202102.0362.v1 Abstract Polyamines are nitrogen-rich polycationic ubiquitous bioactive molecules with diverse evolutionary-conserved functions. Their activity interferes with numerous genes' expression resulting in cell proliferation and signaling modulation. The intracellular levels of polyamines are precisely controlled by evolutionary-conserved machinery. Their transient synthesis is induced by heat stress, radiation, and other traumatic stimuli in a process termed the polyamine stress response (PSR). Notably, polyamine levels decline gradually with age; and external supplementation improves lifespan in model organisms. This corresponds to cytoprotective and reactive oxygen species scavenging properties of polyamines. Paradoxically, age-associated neurodegenerative disorders are characterized by an upsurge in polyamine levels, indicating polyamine pleiotropic, adaptive, and pathogenic roles. Specifically, arginase overactivation and arginine brain deprivation have been shown to play an important role in Alzheimer’s disease (AD) pathogenesis. Here, we assert that a universal short-term PSR associated with acute stimuli is beneficial for survival. -

The Impact of Glutamine Supplementation on the Symptoms
Chen et al. Molecular Neurodegeneration (2016) 11:60 DOI 10.1186/s13024-016-0127-y RESEARCH ARTICLE Open Access The impact of glutamine supplementation on the symptoms of ataxia-telangiectasia: a preclinical assessment Jianmin Chen1* , Yanping Chen1, Graham Vail1, Heiman Chow2, Yang Zhang2, Lauren Louie1, Jiali Li1,3, Ronald P. Hart1, Mark R. Plummer1 and Karl Herrup1,2 Abstract Background: Our previous studies of Alzheimer’s disease (AD) suggested that glutamine broadly improves cellular readiness to respond to stress and acts as a neuroprotectant both in vitro and in AD mouse models. We now expand our studies to a second neurodegenerative disease, ataxia-telangiectasia (A-T). Unlike AD, where clinically significant cognitive decline does not typically occur before age 65, A-T symptoms appear in early childhood and are caused exclusively by mutations in the ATM (A-T mutated) gene. Results: Genetically ATM-deficient mice and wild type littermates were maintained with or without 4 % glutamine in their drinking water for several weeks. In ATM mutants, glutamine supplementation restored serum glutamine and glucose levels and reduced body weight loss. Lost neurophysiological function assessed through the magnitude of hippocampal long term potentiation was significantly restored. Glutamine supplemented mice also showed reduced thymus pathology and, remarkably, a full one-third extension of lifespan. In vitro assays revealed that ATM-deficient cells are more sensitive to glutamine deprivation, while supra-molar glutamine (8 mM) partially rescued the reduction of BDNF expression and HDAC4 nuclear translocation of genetically mutant Atm−/− neurons. Analysis of microarray data suggested that glutamine metabolism is significantly altered in human A-T brains as well. -
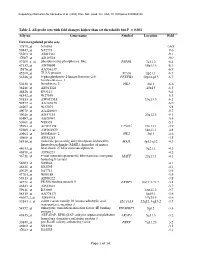
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Structure and Function of Metallohydrolases in the Arginase- Deacetylase Family
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2016 Structure and Function of Metallohydrolases in the Arginase- Deacetylase Family Yang Hai University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons Recommended Citation Hai, Yang, "Structure and Function of Metallohydrolases in the Arginase-Deacetylase Family" (2016). Publicly Accessible Penn Dissertations. 1753. https://repository.upenn.edu/edissertations/1753 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1753 For more information, please contact [email protected]. Structure and Function of Metallohydrolases in the Arginase-Deacetylase Family Abstract Arginases and deacetylases are metallohydrolases that catalyze two distinct chemical transformations. The arginases catalyze the hydrolysis of the guanidinium group of arginine by using a hydroxide ion 2+ 2+ bridging the binuclear manganese cluster (Mn A-Mn B) for nucleophilic attack. The deacetylases catalyze the hydrolysis of amide bonds by using a mononuclear Zn2+-ion activated water molecule as the nucleophile. Despite the diverse functions, metallohydrolases of the arginase-deacetylase superfamily 2+ share the same characteristic α/β hydrolase core fold and a conserved metal binding site (the Mn B site in arginase corresponds to the catalytic Zn2+ site in deacetylase) which is essential for catalysis in both enzymes. We report crystal structure of formiminoglutamase from the parasitic protozoan Trypanosoma cruzi and confirm that formiminoglutamase is a Mn2+-requiring hydrolase that belongs to the arginase- deacetylase superfamily. We also report the crystal structure of an arginase-like protein from Trypanosoma brucei (TbARG) with unknown function. Although its biological role remains enigmatic, the 2+ evolutionarily more conserved Mn B site can be readily restored in TbARG through side-directed mutagenesis.