Alzheimer's Disease As a Chronic Maladaptive Polyamine Stress
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Injury by Mechanical Ventilation Gene Transcription and Promotion Of
Modulation of Lipopolysaccharide-Induced Gene Transcription and Promotion of Lung Injury by Mechanical Ventilation This information is current as William A. Altemeier, Gustavo Matute-Bello, Sina A. of September 29, 2021. Gharib, Robb W. Glenny, Thomas R. Martin and W. Conrad Liles J Immunol 2005; 175:3369-3376; ; doi: 10.4049/jimmunol.175.5.3369 http://www.jimmunol.org/content/175/5/3369 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2005/08/23/175.5.3369.DC1 Material http://www.jimmunol.org/ References This article cites 37 articles, 7 of which you can access for free at: http://www.jimmunol.org/content/175/5/3369.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 29, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Modulation of Lipopolysaccharide-Induced Gene Transcription and Promotion of Lung Injury by Mechanical Ventilation1 William A. -

Blimp1 Regulates the Transition of Neonatal to Adult Intestinal Epithelium
UCLA UCLA Previously Published Works Title Blimp1 regulates the transition of neonatal to adult intestinal epithelium. Permalink https://escholarship.org/uc/item/01x184nd Journal Nature communications, 2(1) ISSN 2041-1723 Authors Muncan, Vanesa Heijmans, Jarom Krasinski, Stephen D et al. Publication Date 2011-08-30 DOI 10.1038/ncomms1463 Peer reviewed eScholarship.org Powered by the California Digital Library University of California ARTICLE Received 11 May 2011 | Accepted 28 Jul 2011 | Published 30 Aug 2011 DOI: 10.1038/ncomms1463 Blimp1 regulates the transition of neonatal to adult intestinal epithelium Vanesa Muncan1,2,3, Jarom Heijmans1,2,3, Stephen D. Krasinski4, Nikè V. Büller1,2,3, Manon E. Wildenberg1,2,3, Sander Meisner1, Marijana Radonjic5, Kelly A. Stapleton4, Wout H. Lamers1, Izak Biemond3, Marius A. van den Bergh Weerman6, Dónal O’Carroll7, James C. Hardwick3, Daniel W. Hommes3 & Gijs R. van den Brink1,2,3 In many mammalian species, the intestinal epithelium undergoes major changes that allow a dietary transition from mother’s milk to the adult diet at the end of the suckling period. These complex developmental changes are the result of a genetic programme intrinsic to the gut tube, but its regulators have not been identified. Here we show that transcriptional repressor B lymphocyte-induced maturation protein 1 (Blimp1) is highly expressed in the developing and postnatal intestinal epithelium until the suckling to weaning transition. Intestine-specific deletion of Blimp1 results in growth retardation and excessive neonatal mortality. Mutant mice lack all of the typical epithelial features of the suckling period and are born with features of an adult-like intestine. -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Determining HDAC8 Substrate Specificity by Noah Ariel Wolfson A
Determining HDAC8 substrate specificity by Noah Ariel Wolfson A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biological Chemistry) in the University of Michigan 2014 Doctoral Committee: Professor Carol A. Fierke, Chair Professor Robert S. Fuller Professor Anna K. Mapp Associate Professor Patrick J. O’Brien Associate Professor Raymond C. Trievel Dedication My thesis is dedicated to all my family, mentors, and friends who made getting to this point possible. ii Table of Contents Dedication ....................................................................................................................................... ii List of Figures .............................................................................................................................. viii List of Tables .................................................................................................................................. x List of Appendices ......................................................................................................................... xi Abstract ......................................................................................................................................... xii Chapter 1 HDAC8 substrates: Histones and beyond ...................................................................... 1 Overview ..................................................................................................................................... 1 HDAC introduction -

Parallel Multi-Omics in High-Risk Subjects for the Identification Of
biomolecules Article Parallel Multi-Omics in High-Risk Subjects for the Identification of Integrated Biomarker Signatures of Type 1 Diabetes Oscar Alcazar 1,†, Luis F. Hernandez 1,†, Ernesto S. Nakayasu 2 , Carrie D. Nicora 2, Charles Ansong 2, Michael J. Muehlbauer 3, James R. Bain 3 , Ciara J. Myer 4,5, Sanjoy K. Bhattacharya 4,5, Peter Buchwald 1,6,*,† and Midhat H. Abdulreda 1,4,7,8,*,† 1 Diabetes Research Institute, University of Miami Miller School of Medicine, Miami, FL 33136, USA; [email protected] (O.A.); [email protected] (L.F.H.) 2 Biological Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99354, USA; [email protected] (E.S.N.); [email protected] (C.D.N.); [email protected] (C.A.) 3 Duke Molecular Physiology Institute, Duke University Medical Center, Durham, NC 27701, USA; [email protected] (M.J.M.); [email protected] (J.R.B.) 4 Department of Ophthalmology, University of Miami Miller School of Medicine, Miami, FL 33136, USA; [email protected] (C.J.M.); [email protected] (S.K.B.) 5 Miami Integrative Metabolomics Research Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA 6 Department of Molecular and Cellular Pharmacology, University of Miami Miller School of Medicine, Miami, FL 33136, USA 7 Department of Surgery, University of Miami Miller School of Medicine, Miami, FL 33136, USA 8 Department of Microbiology and Immunology, University of Miami Miller School of Medicine, Citation: Alcazar, O.; Hernandez, Miami, FL 33136, USA L.F.; Nakayasu, E.S.; Nicora, C.D.; * Correspondence: [email protected] (P.B.); [email protected] (M.H.A.) Ansong, C.; Muehlbauer, M.J.; Bain, † These authors contributed equally. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

The Impact of Transcription Factor Prospero Homeobox 1 on the Regulation of Thyroid Cancer Malignancy
International Journal of Molecular Sciences Review The Impact of Transcription Factor Prospero Homeobox 1 on the Regulation of Thyroid Cancer Malignancy Magdalena Rudzi ´nska 1,2 and Barbara Czarnocka 1,* 1 Department of Biochemistry and Molecular Biology, Centre of Postgraduate Medical Education, 01-813 Warsaw, Poland; [email protected] 2 Institute of Molecular Medicine, Sechenov First Moscow State Medical University, 119991 Moscow, Russia * Correspondence: [email protected]; Tel.: +48-225693812; Fax: +48-225693712 Received: 7 April 2020; Accepted: 30 April 2020; Published: 2 May 2020 Abstract: Transcription factor Prospero homeobox 1 (PROX1) is continuously expressed in the lymphatic endothelial cells, playing an essential role in their differentiation. Many reports have shown that PROX1 is implicated in cancer development and acts as an oncoprotein or suppressor in a tissue-dependent manner. Additionally, the PROX1 expression in many types of tumors has prognostic significance and is associated with patient outcomes. In our previous experimental studies, we showed that PROX1 is present in the thyroid cancer (THC) cells of different origins and has a high impact on follicular thyroid cancer (FTC) phenotypes, regulating migration, invasion, focal adhesion, cytoskeleton reorganization, and angiogenesis. Herein, we discuss the PROX1 transcript and protein structures, the expression pattern of PROX1 in THC specimens, and its epigenetic regulation. Next, we emphasize the biological processes and genes regulated by PROX1 in CGTH-W-1 cells, derived from squamous cell carcinoma of the thyroid gland. Finally, we discuss the interaction of PROX1 with other lymphatic factors. In our review, we aimed to highlight the importance of vascular molecules in cancer development and provide an update on the functionality of PROX1 in THC biology regulation. -

The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors After Cerebral Ischemia: Facts and Hypotheses
International Journal of Molecular Sciences Review The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses Svetlana Demyanenko 1,* and Svetlana Sharifulina 1,2 1 Laboratory of Molecular Neurobiology, Academy of Biology and Biotechnology, Southern Federal University, pr. Stachki 194/1, 344090 Rostov-on-Don, Russia; [email protected] 2 Neuroscience Center HiLife, University of Helsinki, Haartmaninkatu 8, P.O. Box 63, 00014 Helsinki, Finland * Correspondence: [email protected]; Tel.: +7-918-5092185; Fax: +7-863-2230837 Abstract: Histone deacetylase (HDAC) and histone acetyltransferase (HAT) regulate transcription and the most important functions of cells by acetylating/deacetylating histones and non-histone proteins. These proteins are involved in cell survival and death, replication, DNA repair, the cell cycle, and cell responses to stress and aging. HDAC/HAT balance in cells affects gene expression and cell signaling. There are very few studies on the effects of stroke on non-histone protein acetylation/deacetylation in brain cells. HDAC inhibitors have been shown to be effective in protecting the brain from ischemic damage. However, the role of different HDAC isoforms in the survival and death of brain cells after stroke is still controversial. HAT/HDAC activity depends on the acetylation site and the acetylation/deacetylation of the main proteins (c-Myc, E2F1, p53, Citation: Demyanenko, S.; ERK1/2, Akt) considered in this review, that are involved in the regulation of cell fate decisions. Sharifulina, S. The Role of Post-Translational Acetylation and Our review aims to analyze the possible role of the acetylation/deacetylation of transcription factors Deacetylation of Signaling Proteins and signaling proteins involved in the regulation of survival and death in cerebral ischemia. -

The Impact of Glutamine Supplementation on the Symptoms
Chen et al. Molecular Neurodegeneration (2016) 11:60 DOI 10.1186/s13024-016-0127-y RESEARCH ARTICLE Open Access The impact of glutamine supplementation on the symptoms of ataxia-telangiectasia: a preclinical assessment Jianmin Chen1* , Yanping Chen1, Graham Vail1, Heiman Chow2, Yang Zhang2, Lauren Louie1, Jiali Li1,3, Ronald P. Hart1, Mark R. Plummer1 and Karl Herrup1,2 Abstract Background: Our previous studies of Alzheimer’s disease (AD) suggested that glutamine broadly improves cellular readiness to respond to stress and acts as a neuroprotectant both in vitro and in AD mouse models. We now expand our studies to a second neurodegenerative disease, ataxia-telangiectasia (A-T). Unlike AD, where clinically significant cognitive decline does not typically occur before age 65, A-T symptoms appear in early childhood and are caused exclusively by mutations in the ATM (A-T mutated) gene. Results: Genetically ATM-deficient mice and wild type littermates were maintained with or without 4 % glutamine in their drinking water for several weeks. In ATM mutants, glutamine supplementation restored serum glutamine and glucose levels and reduced body weight loss. Lost neurophysiological function assessed through the magnitude of hippocampal long term potentiation was significantly restored. Glutamine supplemented mice also showed reduced thymus pathology and, remarkably, a full one-third extension of lifespan. In vitro assays revealed that ATM-deficient cells are more sensitive to glutamine deprivation, while supra-molar glutamine (8 mM) partially rescued the reduction of BDNF expression and HDAC4 nuclear translocation of genetically mutant Atm−/− neurons. Analysis of microarray data suggested that glutamine metabolism is significantly altered in human A-T brains as well. -
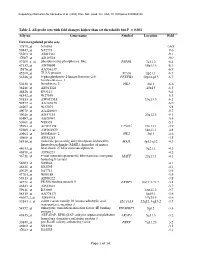
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Structure and Function of Metallohydrolases in the Arginase- Deacetylase Family
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2016 Structure and Function of Metallohydrolases in the Arginase- Deacetylase Family Yang Hai University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons Recommended Citation Hai, Yang, "Structure and Function of Metallohydrolases in the Arginase-Deacetylase Family" (2016). Publicly Accessible Penn Dissertations. 1753. https://repository.upenn.edu/edissertations/1753 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1753 For more information, please contact [email protected]. Structure and Function of Metallohydrolases in the Arginase-Deacetylase Family Abstract Arginases and deacetylases are metallohydrolases that catalyze two distinct chemical transformations. The arginases catalyze the hydrolysis of the guanidinium group of arginine by using a hydroxide ion 2+ 2+ bridging the binuclear manganese cluster (Mn A-Mn B) for nucleophilic attack. The deacetylases catalyze the hydrolysis of amide bonds by using a mononuclear Zn2+-ion activated water molecule as the nucleophile. Despite the diverse functions, metallohydrolases of the arginase-deacetylase superfamily 2+ share the same characteristic α/β hydrolase core fold and a conserved metal binding site (the Mn B site in arginase corresponds to the catalytic Zn2+ site in deacetylase) which is essential for catalysis in both enzymes. We report crystal structure of formiminoglutamase from the parasitic protozoan Trypanosoma cruzi and confirm that formiminoglutamase is a Mn2+-requiring hydrolase that belongs to the arginase- deacetylase superfamily. We also report the crystal structure of an arginase-like protein from Trypanosoma brucei (TbARG) with unknown function. Although its biological role remains enigmatic, the 2+ evolutionarily more conserved Mn B site can be readily restored in TbARG through side-directed mutagenesis.