The Impact of Glutamine Supplementation on the Symptoms
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Determining HDAC8 Substrate Specificity by Noah Ariel Wolfson A
Determining HDAC8 substrate specificity by Noah Ariel Wolfson A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biological Chemistry) in the University of Michigan 2014 Doctoral Committee: Professor Carol A. Fierke, Chair Professor Robert S. Fuller Professor Anna K. Mapp Associate Professor Patrick J. O’Brien Associate Professor Raymond C. Trievel Dedication My thesis is dedicated to all my family, mentors, and friends who made getting to this point possible. ii Table of Contents Dedication ....................................................................................................................................... ii List of Figures .............................................................................................................................. viii List of Tables .................................................................................................................................. x List of Appendices ......................................................................................................................... xi Abstract ......................................................................................................................................... xii Chapter 1 HDAC8 substrates: Histones and beyond ...................................................................... 1 Overview ..................................................................................................................................... 1 HDAC introduction -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors After Cerebral Ischemia: Facts and Hypotheses
International Journal of Molecular Sciences Review The Role of Post-Translational Acetylation and Deacetylation of Signaling Proteins and Transcription Factors after Cerebral Ischemia: Facts and Hypotheses Svetlana Demyanenko 1,* and Svetlana Sharifulina 1,2 1 Laboratory of Molecular Neurobiology, Academy of Biology and Biotechnology, Southern Federal University, pr. Stachki 194/1, 344090 Rostov-on-Don, Russia; [email protected] 2 Neuroscience Center HiLife, University of Helsinki, Haartmaninkatu 8, P.O. Box 63, 00014 Helsinki, Finland * Correspondence: [email protected]; Tel.: +7-918-5092185; Fax: +7-863-2230837 Abstract: Histone deacetylase (HDAC) and histone acetyltransferase (HAT) regulate transcription and the most important functions of cells by acetylating/deacetylating histones and non-histone proteins. These proteins are involved in cell survival and death, replication, DNA repair, the cell cycle, and cell responses to stress and aging. HDAC/HAT balance in cells affects gene expression and cell signaling. There are very few studies on the effects of stroke on non-histone protein acetylation/deacetylation in brain cells. HDAC inhibitors have been shown to be effective in protecting the brain from ischemic damage. However, the role of different HDAC isoforms in the survival and death of brain cells after stroke is still controversial. HAT/HDAC activity depends on the acetylation site and the acetylation/deacetylation of the main proteins (c-Myc, E2F1, p53, Citation: Demyanenko, S.; ERK1/2, Akt) considered in this review, that are involved in the regulation of cell fate decisions. Sharifulina, S. The Role of Post-Translational Acetylation and Our review aims to analyze the possible role of the acetylation/deacetylation of transcription factors Deacetylation of Signaling Proteins and signaling proteins involved in the regulation of survival and death in cerebral ischemia. -

Alzheimer's Disease As a Chronic Maladaptive Polyamine Stress
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 17 February 2021 doi:10.20944/preprints202102.0362.v1 Alzheimer’s disease as a chronic maladaptive polyamine stress response Review Baruh Polis1, David Karasik2,3, Abraham O. Samson1 1 Drug Discovery Laboratory, The Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel. 2 Hebrew SeniorLife, Hinda and Arthur Marcus Institute for Aging Research, Boston, MA, 02131, USA 3 Musculoskeletal Genetics Laboratory, The Azrieli Faculty of Medicine, Bar-Ilan University, Safed, 1311502, Israel. Correspondence to Baruh Polis [email protected] Keywords: polyamines; arginase; senescence; aging; neurodegeneration. 1 © 2021 by the author(s). Distributed under a Creative Commons CC BY license. Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 17 February 2021 doi:10.20944/preprints202102.0362.v1 Abstract Polyamines are nitrogen-rich polycationic ubiquitous bioactive molecules with diverse evolutionary-conserved functions. Their activity interferes with numerous genes' expression resulting in cell proliferation and signaling modulation. The intracellular levels of polyamines are precisely controlled by evolutionary-conserved machinery. Their transient synthesis is induced by heat stress, radiation, and other traumatic stimuli in a process termed the polyamine stress response (PSR). Notably, polyamine levels decline gradually with age; and external supplementation improves lifespan in model organisms. This corresponds to cytoprotective and reactive oxygen species scavenging properties of polyamines. Paradoxically, age-associated neurodegenerative disorders are characterized by an upsurge in polyamine levels, indicating polyamine pleiotropic, adaptive, and pathogenic roles. Specifically, arginase overactivation and arginine brain deprivation have been shown to play an important role in Alzheimer’s disease (AD) pathogenesis. Here, we assert that a universal short-term PSR associated with acute stimuli is beneficial for survival. -
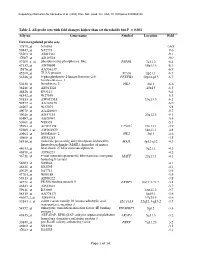
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

Structure and Function of Metallohydrolases in the Arginase- Deacetylase Family
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2016 Structure and Function of Metallohydrolases in the Arginase- Deacetylase Family Yang Hai University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons Recommended Citation Hai, Yang, "Structure and Function of Metallohydrolases in the Arginase-Deacetylase Family" (2016). Publicly Accessible Penn Dissertations. 1753. https://repository.upenn.edu/edissertations/1753 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1753 For more information, please contact [email protected]. Structure and Function of Metallohydrolases in the Arginase-Deacetylase Family Abstract Arginases and deacetylases are metallohydrolases that catalyze two distinct chemical transformations. The arginases catalyze the hydrolysis of the guanidinium group of arginine by using a hydroxide ion 2+ 2+ bridging the binuclear manganese cluster (Mn A-Mn B) for nucleophilic attack. The deacetylases catalyze the hydrolysis of amide bonds by using a mononuclear Zn2+-ion activated water molecule as the nucleophile. Despite the diverse functions, metallohydrolases of the arginase-deacetylase superfamily 2+ share the same characteristic α/β hydrolase core fold and a conserved metal binding site (the Mn B site in arginase corresponds to the catalytic Zn2+ site in deacetylase) which is essential for catalysis in both enzymes. We report crystal structure of formiminoglutamase from the parasitic protozoan Trypanosoma cruzi and confirm that formiminoglutamase is a Mn2+-requiring hydrolase that belongs to the arginase- deacetylase superfamily. We also report the crystal structure of an arginase-like protein from Trypanosoma brucei (TbARG) with unknown function. Although its biological role remains enigmatic, the 2+ evolutionarily more conserved Mn B site can be readily restored in TbARG through side-directed mutagenesis. -

Role of Histone Deacetylases in Gene Expression and RNA Splicing
Role of Histone Deacetylases in Gene Expression and RNA Splicing by Dilshad Hussain Khan A Thesis submitted to the Faculty of Graduate Studies of The University of Manitoba In partial fulfillment of the requirements of the degree of Doctor of Philosophy Department of Biochemistry and Medical Genetics University of Manitoba Winnipeg, Manitoba, Canada Copyright 2013 by Dilshad Hussain Khan Thesis Abstract Histone deacetylases (HDAC) 1 and 2 play crucial role in chromatin remodeling and gene expression regimes, as part of multiprotein corepressor complexes. Protein kinase CK2-driven phosphorylation of HDAC1 and 2 regulates their catalytic activities and is required to form the corepressor complexes. Phosphorylation-mediated differential distributions of HDAC1 and 2 complexes in regulatory and coding regions of transcribed genes catalyze the dynamic protein acetylation of histones and other proteins, thereby influence gene expression. During mitosis, highly phosphorylated HDAC1 and 2 heterodimers dissociate and displace from mitotic chromosomes. Our goal was to identify the kinase involved in mitotic phosphorylation of HDAC1 and 2. We postulated that CK2-mediated increased phosphorylation of HDAC1 and 2 leads to dissociation of the heterodimers, and, the mitotic chromosomal exclusions of HDAC1 and 2 are largely due to the displacement of HDAC-associated proteins and transcription factors, which recruit HDACs, from chromosomes during mitosis. We further explored the role of un- or monomodified HDAC1 and 2 complexes in immediate-early genes (IEGs), FOSL1 (FOS-like antigen-1) and MCL1 (Myeloid cell leukemia-1), regulation. Dynamic histone acetylation is an important regulator of these genes that are overexpressed in a number of diseases and cancers. -

Characterizing the Roles of Exit Tunnel Residues in Ligand Binding
CHARACTERIZING THE ROLES OF EXIT TUNNEL RESIDUES IN LIGAND BINDING AND CATALYSIS OF HISTONE DEACETYLASE-8 A Thesis Submitted to the Graduate Faculty of the North Dakota State University of Agriculture and Applied Science By Ruchi Gupta In Partial Fulfillment for the Degree of MASTER OF SCIENCE Major Department: Chemistry and Biochemistry March 2014 Fargo, North Dakota North Dakota State University Graduate School Title Characterizing the Roles of Exit Tunnel Residues in Ligand Binding and Catalysis of Histone Deacetylase-8 By Ruchi Gupta The Supervisory Committee certifies that this disquisition complies with North Dakota State University’s regulations and meets the accepted standards for the degree of MASTER OF SCIENCE SUPERVISORY COMMITTEE: Dr. D.K. Srivastava Chair Dr. Gregory Cook Dr. Stuart Haring Dr. Jane Schuh Approved: 03/26/2014 Gregory Cook Date Department Chair ABSTRACT Histone deacetylases are an important class of enzymes that catalyze the hydrolysis of acetyl-L-lysine side chains in histone and non-histone proteins to yield L-lysine and acetate, effecting the epigenetic regulation of gene expression. In addition to the active site pocket, the enzyme harbors an internal cavity for the release of acetate by-product. To probe the role of highly conserved amino acid residues lining this exit tunnel, site-directed alanine substitutions were made at tyrosine-18, tyrosine-20 and histidine-42 positions. These mutants were characterized by various biochemical and biophysical techniques to define the effect of mutations on ligand binding and catalysis of the enzyme. The mutations altered the catalytic activity of HDAC8 significantly. Y18A mutation dramatically impaired the structural-functional aspects of the enzymatic reaction. -

The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis
J Lipid Atheroscler. 2020 Sep;9(3):419-434 Journal of https://doi.org/10.12997/jla.2020.9.3.419 Lipid and pISSN 2287-2892·eISSN 2288-2561 Atherosclerosis Review The Key Role of DNA Methylation and Histone Acetylation in Epigenetics of Atherosclerosis Han-Teo Lee ,1,2,* Sanghyeon Oh ,1,2,* Du Hyun Ro ,1,2,3,* Hyerin Yoo ,1,2 Yoo-Wook Kwon 4,5 1Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science, Seoul National University, Seoul, Korea 2Interdisciplinary Program in Stem Cell Biology, Graduate School of Medicine, Seoul National University, Seoul, Korea 3Department of Orthopedic Surgery, Seoul National University Hospital, Seoul, Korea 4Strategic Center of Cell and Bio Therapy for Heart, Diabetes & Cancer, Biomedical Research Institute, Seoul National University Hospital, Seoul, Korea Received: May 8, 2020 5Department of Medicine, College of Medicine, Seoul National University, Seoul, Korea Revised: Sep 14, 2020 Accepted: Sep 15, 2020 Correspondence to ABSTRACT Yoo-Wook Kwon Biomedical Research Institute, Seoul National Atherosclerosis, which is the most common chronic disease of the coronary artery, constitutes University Hospital, 103 Daehak-ro, Jongno- a vascular pathology induced by inflammation and plaque accumulation within arterial vessel gu, Seoul 03080, Korea. E-mail: [email protected] walls. Both DNA methylation and histone modifications are epigenetic changes relevant for atherosclerosis. Recent studies have shown that the DNA methylation and histone *Han-Teo Lee, Sanghyeon Oh and Du Hyun Ro modification systems are closely interrelated and mechanically dependent on each other. contributed equally to this work. Herein, we explore the functional linkage between these systems, with a particular emphasis Copyright © 2020 The Korean Society of Lipid on several recent findings suggesting that histone acetylation can help in targeting DNA and Atherosclerosis. -

Characterization of Histone Deacetylase Expression Within in Vitro and in Vivo Bladder Cancer Model Systems
International Journal of Molecular Sciences Article Characterization of Histone Deacetylase Expression Within In Vitro and In Vivo Bladder Cancer Model Systems Jenna M. Buckwalter 1,2,*, Wilson Chan 2, Lauren Shuman 1,2, Thomas Wildermuth 1,2, Justine Ellis-Mohl 1,2, Vonn Walter 3,4, Joshua I. Warrick 1,2, Xue-Ru Wu 5, Matt Kaag 2, Jay D. Raman 2 and David J. DeGraff 1,2 1 Department of Pathology and Laboratory Medicine, Pennsylvania State University College of Medicine, Hershey, PA 17033, USA; [email protected] (L.S.); [email protected] (T.W.); [email protected] (J.E.-M.); [email protected] (J.I.W.); ddegraff@pennstatehealth.psu.edu (D.J.D.) 2 Department of Surgery, Division of Urology, Pennsylvania State University College of Medicine, Hershey, PA 17033, USA; [email protected] (W.C.); [email protected] (M.K.); [email protected] (J.D.R.) 3 Department of Public Health Sciences, Pennsylvania State University College of Medicine, Hershey, PA 17033, USA; [email protected] 4 Department of Biochemistry and Molecular Biology, Pennsylvania State University College of Medicine, Hershey, PA 17033, USA 5 Departments of Urology and Pathology, New York University, New York, NY 10010, USA; [email protected] * Correspondence: [email protected]; Tel.: +717-531-0003 Received: 7 May 2019; Accepted: 22 May 2019; Published: 27 May 2019 Abstract: Epigenetic aberrations are prominent in bladder cancer (BC) and contribute to disease pathogenesis. We characterized histone deacetylase (HDAC) expression, a family of deacetylation enzymes, in both in vitro and in vivo BC model systems and analyzed expression data from The Cancer Genome Atlas (TCGA). -

Dissertation / Doctoral Thesis
DISSERTATION / DOCTORAL THESIS Titel der Dissertation /Title of the Doctoral Thesis A robust and high-throughput Assay to study Somatic Hypermutation unveiling the role of 14-3-3 Adaptor Proteins verfasst von / submitted by Dott.ssa.mag. Marialaura Mastrovito angestrebter akademischer Grad / in partial fulfilment of the requirements for the degree of Doctor of Philosophy (PhD) Wien, 2018 / Vienna, 2018 Studienkennzahl lt. Studienblatt / A 794 685 490 degree programme code as it appears on the student record sheet: Dissertationsgebiet lt. Studienblatt / Molekulare Biologie field of study as it appears on the student record sheet: Betreut von / Supervisor: Rushad Pavri, PhD Table of contents TABLE OF CONTENTS Acknowledgments .............................................................................................................. 5 Abstract .............................................................................................................................. 6 Zusammenfassung ............................................................................................................. 7 List of abbreviations .......................................................................................................... 9 1. INTRODUCTION ....................................................................................................... 11 1.1 Antibody diversification ........................................................................................................... 11 1.2 Molecular mechanism of SHM and CSR ............................................................................ -

Unveiling Human Non-Random Genome Editing Mechanisms Activated in Response to Chronic Environmental Changes
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 19 July 2020 Unveiling human non-random genome editing mechanisms activated in response to chronic environmental changes. I. Where might these mechanisms come from? Loris Zamai Department of Biomolecular Sciences, University of Urbino Carlo Bo; Urbino; National Institute for Nuclear Physics (INFN)-Gran Sasso National Laboratory (LNGS), Assergi, L'Aquila, Italy. Corresponding author: Loris Zamai, Department of Biomolecular Sciences, via Ca’ le Suore 2, University of Urbino Carlo Bo, 61029 Urbino, Italy. Tel. (+39) 0722 304319; fax (+39) 0722 304319; e-mail: [email protected] Running Headline: Non-random genome editing mechanisms Key words: environment, virus, pollutant, evolution, exaptation, stem cells, transposons, LINE-1, Alu, mutagenic enzymes, APOBEC, ADAR, ORF2p, cancer, Eco-Evo-Devo, symbiosis, ecological genomics, environmental stress, genetic recombination, biological plasticity, hypermutation, epigenetics, fractal systems, natural selection. List of Abbreviations single nucleotide polymorphisms, (SNP); copy number variations, (CNV); stress induced mutagenesis, (SIM); ecological evolutionary developmental biology, (Eco-Evo-Devo); somatic hypermutation, (SHM); immunoglobulin, (Ig); activation-induced deaminase (AID); CRISPR, (clustered, regularly interspaced, short, palindromic repeats); Cas, (CRISPR-associated); CRISPR RNAs, (crRNAs); Cascade, (CRISPR-associated complex for antiviral defense); apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like, (APOBEC);