Virome Profiling of Rodents in Xinjiang Uygur Autonomous Region, China
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Keeping Friends Close, and Their Oil Closer: Rethinking the Role of The
Keeping Friends Close, and Their Oil Closer: Rethinking the Role of the Shanghai Cooperation Organization in China’s Strive for Energy Security in Kazakhstan By Milos Popovic Submitted to Central European University Department of International Relations and European Studies In partial fulfillment of the requirements for the degree of Master of Arts in International Relations and European Studies Supervisor: Professor Matteo Fumagalli Word Count: 17, 201 CEU eTD Collection Budapest, Hungary June, 2010 ABSTRACT It is generally acknowledged that Beijing’s bilateral oil dealings pertaining to the construction of the Atyrau-Alashankou pipeline comprise the backbone of China’s strive for energy security in Kazakhstan. Against the backdrop of a widespread scholarly claim that the Shanghai Cooperation Organization (SCO) plays no role in this endeavor, this thesis argues that Beijing acts as a security-seeker to bind both Kazakhstan and Russia into energy cooperation within the organization. Acting as a regional forum through which China channels and reinforces its oil dealings, I argue that the SCO corrects the pitfalls of a bilateral approach which elicits the counter-balancing of Chinese activities by Astana and Moscow who are concerned with the distribution of gains. Putting to a test differing hypothesis by rationalist IR theories, I find that the SCO approach enables China to assure both actors about its benign intentions and maximize gains on a bilateral level as expected by defensive neorealism. CEU eTD Collection i ACKNOWLEDGEMENTS My immense love and gratitude belongs to my parents and brother who wholeheartedly supported me during the course of the whole academic year giving me the strength to endure amid hard times. -

Master's Degree Programme
Master’s Degree Programme In Languages, Economics and Institutions of Asia and North Africa “D.M. 270/2004” Final Thesis Chongqing-Xinjiang-Europe International Railway: problems and challenges of the first direct rail connection between China and Europe Supervisor Ch. Prof. Riccardo Renzo Cavalieri Assistant supervisor Ch. Prof. Daniele Brombal Graduand Irene Tambellini Matriculation Number 966550 Academic Year 2017 / 2018 前言 自 1978 年邓小平主席改革开放以来,中国开始了现代化以及改变了国 家政治经济格局的改革进程。中国打开了面向世界的大门,成为全球 化进程中的一个重要角色,并且开始建立与其他国家的经济合作伙伴 关系。事实上,上个世纪九十年代末中国和欧洲的经济合作就已开始 发展。使他们的合作伙伴关系更加紧密的阶段有多个,2001 年中国加 入了世界贸易组织,2003 年中国和欧盟签署了“中-欧”战略合作伙 伴关系,并且在 2013 年采纳并签署了批准双方全方位合作的“欧-中 2020 年战略合作议程”。今天,欧洲成为中国的第一大贸易伙伴,而 中国成为欧洲的第二大贸易伙伴。因此自 1978 年以来,中国经济经历 了指数性增长并且中国在国际事务中的参与度也得到提升。这项长久 进程在 2013 年习近平主席提出一带一路倡议时达到高峰,一个宏伟的 项目被计划出来以建立亚洲,欧洲以及非洲国家之间的政治经济网络。 这个项目又两部分构成:陆地部分被称为丝绸之路经济带,海上部分 被称为 21 世纪海上丝绸之路。所以,连接中国和世界的基础设施的建 设代表着一个最重要的可以引领这个战略取得成功的要素。由于这个 原因,沿这条路上的国家之间的沿海和陆地走廊的发展受到了很大的 重视。欧洲国家在一带一路倡议的构架中扮演着重要的角色,并且在 这些国家中项目的数量以及投资都很多,德国代表着那些在中国最重 要的合作伙伴。的确,德国是中国与欧盟国家最大的贸易伙伴,并且 中国也代表着德国的第二大出口市场。因此,德国在一带一路倡议中 也具有重要地位并且中国与德国诸多城市之间建立的铁路联接也是其 重要角色的证明。这篇文章的目的是分析建立在中国与欧洲之间的第 一条直线铁路连接,评估其竞争力,尤其是与其他交通方式做比较。 1 这条铁路线的名称是重庆-新疆-欧洲国际铁路或渝新欧国际铁路,它 以重庆作为起始站到达德国的杜伊斯堡,途径哈萨克斯坦,俄罗斯, 白俄罗斯以及波兰。 在第一章中,我将聚焦于重庆市并且描述使得中 国政府建立这座直辖市的经济背景。自上世纪九十年代末起,中国开 始集中于国内的发展,尤其是对于一直以来落后于沿海省份的内部地 区的发展。由于这个原因,中央以引导这些地区的经济扩展为目标指 出了一些增长极点. 直辖市重庆便是这些增长极点中的一个,自 1999 年西部大开发战略开始以来,重庆扮演了中国经济政策的一个重要角 色。我将会称述西部大开发战略背后的动机以及中央政府出于发展内 部地区所采取的政策。重庆作为西部省份发展的必不可少的角色,将 被在西部大开发以及一带一路倡议中进行研究。因此,本文将表述重 庆转型为一个重要经济中心的过程。在第二章中,我将考察一带一路 倡议前后中国和欧洲建立的沿海和内陆联系. -

Forest Dormouse (Dryomys Nitedula, Rodentia, Gliridae) – a Highly Contagious Rodent in Relation to Zoonotic Diseases
FORESTRY IDEAS, 2020, vol. 26, No 1 (59): 262–269 FOREST DORMOUSE (DRYOMYS NITEDULA, RODENTIA, GLIRIDAE) – A HIGHLY CONTAGIOUS RODENT IN RELATION TO ZOONOTIC DISEASES Alexey Andreychev1* and Ekaterina Boyarova2 1Department of Zoology, National Research Mordovia State University, Saransk 430005, Russia. *E-mail: [email protected] 2Center for Hygiene and Epidemiology in the Republic of Mordovia, Saransk 430030, 1a Dal’naya Str., Russia. Received: 22 March 2020 Accepted: 23 May 2020 Abstract Republic of Mordovia in Russia is a historical focus for hemorrhagic fever with renal syndrome and tularemia. This study aimed at assessing the current status of these foci by studying their rodent reservoirs. Among the small rodents in Mordovia, the red bank vole, the common vole and the house mouse play an important role as carriers of zoonotic diseases. However, it is neces- sary to take into account the role of such а small species as forest dormouse (Dryomys nitedula), which has a high percentage of infection. Of all examined dormice, 66.7 % were found to have antigens of hemorrhagic fever with renal syndrome viral pathogen, and 33.3 % - to have antigens of tularemia pathogen. Only one specimen (16.7 %) was found to have no antigens of zoonotic diseases. Our study concluded that the forest dormouse in the Republic of Mordovia was incrim- inated as a pathogen reservoir causing infectious diseases in human. Key words: hemorrhagic fever, Mordovia, rodents, tularemia. Introduction gion of Middle Volga (Ulyanovsk region) (Nafeev et al. 2009). Small mammals are Hemorrhagic fever with renal syndrome the most incriminated reservoirs for viral (HFRS) is the most common natural ro- pathogen agents causing human HFRS dent borne disease of viral etiology in the in the natural environments (Tsvirko and Republic of Mordovia. -
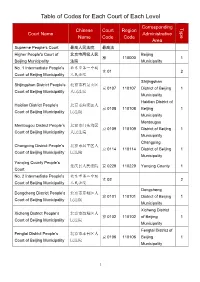
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

Seasonal Changes in Tawny Owl (Strix Aluco) Diet in an Oak Forest in Eastern Ukraine
Turkish Journal of Zoology Turk J Zool (2017) 41: 130-137 http://journals.tubitak.gov.tr/zoology/ © TÜBİTAK Research Article doi:10.3906/zoo-1509-43 Seasonal changes in Tawny Owl (Strix aluco) diet in an oak forest in Eastern Ukraine 1, 2 Yehor YATSIUK *, Yuliya FILATOVA 1 National Park “Gomilshanski Lisy”, Kharkiv region, Ukraine 2 Department of Zoology and Animal Ecology, Faculty of Biology, V.N. Karazin Kharkiv National University, Kharkiv, Ukraine Received: 22.09.2015 Accepted/Published Online: 25.04.2016 Final Version: 25.01.2017 Abstract: We analyzed seasonal changes in Tawny Owl (Strix aluco) diet in a broadleaved forest in Eastern Ukraine over 6 years (2007– 2012). Annual seasons were divided as follows: December–mid-April, April–June, July–early October, and late October–November. In total, 1648 pellets were analyzed. The most important prey was the bank vole (Myodes glareolus) (41.9%), but the yellow-necked mouse (Apodemus flavicollis) (17.8%) dominated in some seasons. According to trapping results, the bank vole was the most abundant rodent species in the study region. The most diverse diet was in late spring and early summer. Small forest mammals constituted the dominant group in all seasons, but in spring and summer their share fell due to the inclusion of birds and the common spadefoot (Pelobates fuscus). Diet was similar in late autumn, before the establishment of snow cover, and in winter. The relative representation of species associated with open spaces increased in winter, especially in years with deep snow cover, which may indicate seasonal changes in the hunting habitats of the Tawny Owl. -

The New Asia-Europe Land Bridge— Current Situation and Future Prospects Xu Shu
Features International Cooperation (part 2) The New Asia-Europe Land Bridge— Current Situation and Future Prospects Xu Shu In September 1990, China’s Bei-jiang Middle East. The line is benefitting the Concept of Line linking Urumqi and Alashankou was development of the regions it serves. New Asia-Europe Land Bridge connected to Kazakstan Railways, At an ASEAN conference in Bangkok in thereby linking Lianyungang and other March 1996, attended by European lead- Regional cooperation and development ports in east China directly by rail with ers, it was stated that Europe and Asia is an important trend in current world Rotterdam, Holland and other European should make a concerted effort to catch affairs. The new economic zone spans ports (Fig. 1). Freight and passenger ser- the favourable tide and redouble their the central land mass of Eurasia from the vices were inaugurated on 1 December efforts to meet the 21st century with new west Pacific coast to the east Atlantic 1992. This New Asia-Europe Land Bridge amicable relations. The meeting called coast. The 10,900-km link is a hugely spans two continents, a link made pos- for increased dialogue, a deepening of important international route uniting over sible by the work of the Chinese govern- economic ties, and increased coopera- forty nations covering 39.7 million km2 ment and ministries, and commercial tion in light of common aspirations and or 26.6% of the world’s land area. The business enterprises. needs. In 1996, the New Asia-Europe population of this area is 2.2 billion, or The completion of the New Land Bridge Land Bridge Railway Regional Economic 36% of the total. -

CAREC Corridor Implementation Progress, Planned Actions, and Support Needs
CAREC Corridor Implementation Progress, Planned Actions, and Support Needs P. R. China Istanbul, Apr. 18th, 2018 Infrastructure Инфраструктура 5 Routes in China's Territory: 5 маршруты в китайсой стороне: (a) Route 1: Hami-Turpan-Urumqi-Kuytun-Jinghe-Alashankou (а)Маршрут 1:Хами-Турфан-Урумчи-Куйтун-Цинхэ-Алашанькоу. (b) Route 2: Hami-Turpan-Urumqi-Kuytun-Jinghe-Khorgos Маршрут 2:Хами-Турфан-Урумчи-Куйтун-Цинхэ-Хоргос (c)Rout 3: Hami-Turpan-Kashi-Torugart (в)Маршрут 3:Хами-Турфан-Кашгар-Тургат (d) Route 4: Hami-Turpan-Karshi-Yierkeshitan (г)Маршрут 4:Хами-Турфан-Кашгар-Иркештам (e) Route 5: Urumqi-Takeshiken (д)Маршрут 5:Урумчи-Такшкин Map of Relevant Routes in China Китайская планировочная схема соответствующих сетей Status of the 5 Routes Конкретно о 5 маршрутах Route 1: Hami-Turpan-Urumqi-Kuytun-Jinghe-Alashankou (1a, 2c) Маршрут 1:Хами-Турфан-Урумчи-Куйтун-Цинхэ-Алашанькоу * Hami-Jinghe Section (G30): Motorway. Xiaocaohu-Urumqi and Urumqi-Kuytun: in upgradation, about 360 km * Участок Хами-Цинхэ (G30),Четырехполосная скоростная автотрасса. Сяосаоху-Урумчи,Урумчи-Куйтун, в процессе реконструтирования, 360 км. * Jinghe-Alashankou (G3018): Motorway under construction; most parts of the existing roads - Grade I Highway * Участок Цинхэ-Алашанькоу (G3018),незавершенная скоростная автотрасса. Часть-первоклассное шоссе. Route 2:Hami-Turpan-Urumqi-Kuytun-Jinghe-Khorgos (1b) Маршрут 2:Хами-Турфан-Урумчи-Куйтун-Цинхэ-Хоргос * Hami-Khorgos (G30): Motorway. Xiaocaohu-Urumqi, Urumqi- Kuytun: in upgradation, about 360 km * Участок Хами-Хоргос(G30), Четырехполосная скоростная а втотрасса. Сяосаоху-Урумчи,Урумчи-Куйтун, в процессе реконструтирования, 360 км. Rout 3: Hami-Turpan-Kashi-Torugart (1c) Маршрут 3:Хами-Турфан-Кашгар-Тургат * Hami-Turpan (G30): Motorway. -

Frontier Politics and Sino-Soviet Relations: a Study of Northwestern Xinjiang, 1949-1963
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Frontier Politics And Sino-Soviet Relations: A Study Of Northwestern Xinjiang, 1949-1963 Sheng Mao University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the History Commons Recommended Citation Mao, Sheng, "Frontier Politics And Sino-Soviet Relations: A Study Of Northwestern Xinjiang, 1949-1963" (2017). Publicly Accessible Penn Dissertations. 2459. https://repository.upenn.edu/edissertations/2459 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2459 For more information, please contact [email protected]. Frontier Politics And Sino-Soviet Relations: A Study Of Northwestern Xinjiang, 1949-1963 Abstract This is an ethnopolitical and diplomatic study of the Three Districts, or the former East Turkestan Republic, in China’s northwest frontier in the 1950s and 1960s. It describes how this Muslim borderland between Central Asia and China became today’s Yili Kazakh Autonomous Prefecture under the Xinjiang Uyghur Autonomous Region. The Three Districts had been in the Soviet sphere of influence since the 1930s and remained so even after the Chinese Communist takeover in October 1949. After the Sino- Soviet split in the late 1950s, Beijing transformed a fragile suzerainty into full sovereignty over this region: the transitional population in Xinjiang was demarcated, border defenses were established, and Soviet consulates were forced to withdraw. As a result, the Three Districts changed from a Soviet frontier to a Chinese one, and Xinjiang’s outward focus moved from Soviet Central Asia to China proper. The largely peaceful integration of Xinjiang into PRC China stands in stark contrast to what occurred in Outer Mongolia and Tibet. -

Disfranchised Under the Name of Zhonghua Minzu Wenjun Zeng
Disfranchised under the name of Zhonghua Minzu Wenjun Zeng Innovative Research in Conflict Analysis NYU IR MA Program Fall 2015 Disfranchised under the name of Zhonghua Minzu 2 Wenjun Zeng New York University Introduction: The origin and development of Zhonghua Minzu (Chinese Nation) China is a country with fifty-six ethnicities with Han being the vast majority. In China, many minorities own their own distinctive history and culture.1 Yet, these 56 ethnicities are usually simplified under the title of Zhonghua Minzu (Chinese national) by the government and Han people under political, social, and cultural realm. Under such identity, all Chinese nationals share the same ethnic origin of being the decedents of Yan, and Huang Emperor, two mythical figures from the ancient Chinese folklore.2 Zhonghua Minzu was created by a Chinese philosopher and revolutionary for articulating the anti-Manchurian identity during the late Qing Dynasty.3 It then remained until the establishment of the People’s Republic of China (PRC), by the Chinese Communist Party (CCP), as a political rhetoric to represent a unified Chinese identity. Minorities in China today assimilated their culture into Han Chinese on different degrees, including their languages, daily attires, social values, and religions. Chinese nation and Han ethnicity are interchangeable under many circumstances. However, even decades after CCP’s governing, Uyghurs, one of the Turkic Sunni Muslim minorities, remains distinct from the mainstream Chinese identity. The province they concentrated in, Xinjiang Uyghur Autonomous Region (XUAR), located in the western periphery of the country as showed in the Map 1. As its Chinese name “Xinjiang”—the “new territory”–implies, Xinjiang only became part of the Chinese administration in Qing Dynasty in 1 Over 90% of the Chinese population is Han. -

The Turkmen Lake Altyn Asyr and Water Resources in Turkmenistan the Handbook of Environmental Chemistry
The Handbook of Environmental Chemistry 28 Series Editors: Damià Barceló · Andrey G. Kostianoy Igor S. Zonn Andrey G. Kostianoy Editors The Turkmen Lake Altyn Asyr and Water Resources in Turkmenistan The Handbook of Environmental Chemistry Founded by Otto Hutzinger Editors-in-Chief: Damia` Barcelo´ l Andrey G. Kostianoy Volume 28 Advisory Board: Jacob de Boer, Philippe Garrigues, Ji-Dong Gu, Kevin C. Jones, Thomas P. Knepper, Alice Newton, Donald L. Sparks The Handbook of Environmental Chemistry Recently Published and Forthcoming Volumes The Turkmen Lake Altyn Asyr and Emerging and Priority Pollutants in Water Resources in Turkmenistan Rivers: Bringing Science into River Volume Editors: I.S. Zonn Management Plans and A.G. Kostianoy Volume Editors: H. Guasch, A. Ginebreda, Vol. 28, 2014 and A. Geiszinger Vol. 19, 2012 Oil Pollution in the Baltic Sea Global Risk-Based Management of Volume Editors: A.G. Kostianoy Chemical Additives I: Production, and O.Yu. Lavrova Usage and Environmental Occurrence Vol. 27, 2014 Volume Editors: B. Bilitewski, R.M. Darbra, and D. Barcelo´ Urban Air Quality in Europe Vol. 18, 2012 Volume Editor: M. Viana Vol. 26, 2013 Polyfluorinated Chemicals and Transformation Products Climate Change and Water Resources Volume Editors: T.P. Knepper Volume Editors: T. Younos and C.A. Grady and F.T. Lange Vol. 25, 2013 Vol. 17, 2012 Emerging Organic Contaminants in Brominated Flame Retardants Sludges: Analysis, Fate and Biological Volume Editors: E. Eljarrat and D. Barcelo´ Treatment Vol. 16, 2011 Volume Editors: T. Vicent, G. Caminal, E. Eljarrat, and D. Barcelo´ Effect-Directed Analysis of Complex Vol. 24, 2013 Environmental Contamination Volume Editor: W. -

Climate System in Northwest China ������������������������������������������������������ 51 Yaning Chen, Baofu Li and Changchun Xu
Water Resources Research in Northwest China Yaning Chen Editor Water Resources Research in Northwest China 1 3 Editor Yaning Chen Xinjiang Institute of Ecology and Geography Chinese Academy of Sciences Xinjiang People’s Republic of China ISBN 978-94-017-8016-2 ISBN 978-94-017-8017-9 (eBook) DOI 10.1007/978-94-017-8017-9 Springer Dordrecht Heidelberg New York London Library of Congress Control Number: 2014930889 © Springer Science+Business Media Dordrecht 2014 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. Exempted from this legal reservation are brief excerpts in connection with reviews or scholarly analysis or material supplied specifically for the purpose of being entered and executed on a computer system, for exclusive use by the purchaser of the work. Duplication of this publication or parts thereof is permitted only under the provisions of the Copyright Law of the Publisher’s location, in its current version, and permission for use must always be obtained from Springer. Permissions for use may be obtained through RightsLink at the Copyright Clearance Center. Violations are liable to prosecution under the respective Copyright Law. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. -

36496 Federal Register / Vol
36496 Federal Register / Vol. 86, No. 130 / Monday, July 12, 2021 / Rules and Regulations compensation is provided solely for the under forty-three entries to the Entity Committee (ERC) to be ‘military end flight training and not the use of the List. These thirty-four entities have been users’ pursuant to § 744.21 of the EAR. aircraft. determined by the U.S. Government to That section imposes additional license The FAA notes that any operator of a be acting contrary to the foreign policy requirements on, and limits the limited category aircraft that holds an interests of the United States and will be availability of, most license exceptions exemption to conduct Living History of listed on the Entity List under the for, exports, reexports, and transfers (in- Flight (LHFE) operations already holds destinations of Canada; People’s country) to listed entities on the MEU the necessary exemption relief to Republic of China (China); Iran; List, as specified in supplement no. 7 to conduct flight training for its flightcrew Lebanon; Netherlands (The part 744 and § 744.21 of the EAR. members. LHFE exemptions grant relief Netherlands); Pakistan; Russia; Entities may be listed on the MEU List Singapore; South Korea; Taiwan; to the extent necessary to allow the under the destinations of Burma, China, exemption holder to operate certain Turkey; the United Arab Emirates Russia, or Venezuela. The license aircraft for the purpose of carrying (UAE); and the United Kingdom. This review policy for each listed entity is persons for compensation or hire for final rule also revises one entry on the identified in the introductory text of living history flight experiences.