Type I Interferons Mediate Pancreatic Toxicities of PERK Inhibition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supporting Information
Supporting Information Celhar et al. 10.1073/pnas.1507052112 SI Materials and Methods using a Nanodrop spectrophotometer (Thermo Fisher Scien- Proteinuria. Proteinuria was assessed using Albustix (Bayer). Al- tific). A TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems) bumin levels in urine were assayed using an Albumin Mouse was used to perform the reverse transcription and quantitative ELISA Kit (Abcam) according to the manufacturer’s instructions; PCR reactions according to the manufacturer’s instructions samples were assayed at a dilution of 1:400. Samples were nor- using TaqMan gene expression assays (Applied Biosystems) to malized for creatinine using a Creatinine (urinary) Colorimetric either Tlr7 (Mm00446590) or the B2m housekeeping gene Assay Kit (Cayman Chemical) according to the manufacturer’s (Mm00437762). Real-time PCR was performed on the 7900H instructions; initial sample dilution of 1:10. fast real-time PCR system and analyzed using SDS 2.4 (Applied Biosystems). Relative mRNA expression was calculated using the Cell Sorting, RNA Isolation, and RT-PCR. Splenic B cells were comparative C method. + − + + t sorted as live CD45 Gr1 B220 CD19 , splenic T cells as live + − + + CD45 Gr1 CD3 CD5 and peritoneal macrophages as live Imaging. Kidney sections from OCT embedded tissue were fixed + − CD45 Gr1 CD11bhiF4/80hi. Sorted cells were centrifuged, re- with 4% paraformaldehyde before permeabilization with acetone suspended in TRIzol (Life Technologies) and stored at −80°. RNA and stained with Phalloidin (AF647) and anti-CD3d (unlabeled was extracted by TRIzol/chloroform and purified with the Qiagen Ab followed by secondary staining with donkey anti-goat Dylight RNeasy Mini purification kit according to the manufacturer’s 550). -

Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis
Virginia Commonwealth University VCU Scholars Compass Theses and Dissertations Graduate School 2010 Molecular Mechanisms Involved Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis Ryan Fassnacht Virginia Commonwealth University Follow this and additional works at: https://scholarscompass.vcu.edu/etd Part of the Physiology Commons © The Author Downloaded from https://scholarscompass.vcu.edu/etd/2246 This Thesis is brought to you for free and open access by the Graduate School at VCU Scholars Compass. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of VCU Scholars Compass. For more information, please contact [email protected]. Ryan C. Fassnacht 2010 All Rights Reserved Molecular Mechanisms Involved in the Interaction Effects of HCV and Ethanol on Liver Cirrhosis A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science at Virginia Commonwealth University. by Ryan Christopher Fassnacht, B.S. Hampden Sydney University, 2005 M.S. Virginia Commonwealth University, 2010 Director: Valeria Mas, Ph.D., Associate Professor of Surgery and Pathology Division of Transplant Department of Surgery Virginia Commonwealth University Richmond, Virginia July 9, 2010 Acknowledgement The Author wishes to thank his family and close friends for their support. He would also like to thank the members of the molecular transplant team for their help and advice. This project would not have been possible with out the help of Dr. Valeria Mas and her endearing -

A Functional C-Terminal TRAF3-Binding Site in MAVS Participates in Positive and Negative Regulation of the IFN Antiviral Response
Cell Research (2011) 21:895-910. © 2011 IBCB, SIBS, CAS All rights reserved 1001-0602/11 $ 32.00 npg ORIGINAL ARTICLE www.nature.com/cr A functional C-terminal TRAF3-binding site in MAVS participates in positive and negative regulation of the IFN antiviral response Suzanne Paz1, 2, Myriam Vilasco3, Steven J Werden1, Meztli Arguello1, Deshanthe Joseph-Pillai1, 2, Tiejun Zhao1, Thi Lien-Anh Nguyen1, Qiang Sun1, Eliane F Meurs3, Rongtuan Lin1, 4, John Hiscott1, 2, 4 1Terry Fox Molecular Oncology Group, Lady Davis Institute, Jewish General Hospital, Montreal, Quebec H3T1E2, Canada; 2Department of Microbiology and Immunology, McGill University Montreal, Quebec H3A 2B4, Canada; 3Department of Virology, Unit of Hepacivirus and Innate Immunity, Pasteur Institute, Paris 75724 France; 4Department of Medicine, McGill University Montreal, Quebec H3A 2B4, Canada Recognition of viral RNA structures by the cytosolic sensor retinoic acid-inducible gene-I (RIG-I) results in the activation of signaling cascades that culminate with the generation of the type I interferon (IFN) antiviral response. Onset of antiviral and inflammatory responses to viral pathogens necessitates the regulated spatiotemporal recruitment of signaling adapters, kinases and transcriptional proteins to the mitochondrial antiviral signaling protein (MAVS). We previously demonstrated that the serine/threonine kinase IKKε is recruited to the C-terminal region of MAVS following Sendai or vesicular stomatitis virus (VSV) infection, mediated by Lys63-linked polyubiquitination of MAVS at Lys500, resulting in inhibition of downstream IFN signaling (Paz et al, Mol Cell Biol, 2009). In this study, we demonstrate that C-terminus of MAVS harbors a novel TRAF3-binding site in the aa450- 468 region of MAVS. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Human Ifnα4 / Ifna4 / Interferon Alpha-4 Protein (Fc Tag)
Human IFNα4 / IFNa4 / Interferon alpha-4 Protein (Fc Tag) Catalog Number: 10336-H01H General Information SDS-PAGE: Gene Name Synonym: IFN-alpha4a; INFA4; MGC142200 Protein Construction: A DNA sequence encoding the mature form of human IFN-α4a (NP_066546.1) (Cys 23-Asp 189) was expressed with the N-terminal fused Fc region of human IgG1. Source: Human Expression Host: HEK293 Cells QC Testing Purity: > 97 % as determined by SDS-PAGE Bio Activity: Protein Description Measured in antiviral assays using WISH human amnion cells infected Interferon, alpha 4 (IFNA4) belongs to the alpha/beta interferon family. Two with vesicular stomatitis virus (VSV). The EC50 for this effect is 0.2-1 variants of IFNA4 (IFNA4a and IFNA4b) are known, which differ from each ng/mL. other by changes in their coding regions at nucleotide positions 220 and 410 and can be distinguished by selective restriction enzyme analysis. Endotoxin: Interferons are produced by macrophages, IFN-alpha have antiviral activities. Interferon stimulates the production of two enzymes: a protein < 1.0 EU per μg of the protein as determined by the LAL method kinase and an oligoadenylate synthetase. IFN-alpha, the first cytokine to be produced by recombinant DNA technology, has emerged as an important Stability: regulator of growth and differentiation, affecting cellular communication and Samples are stable for up to twelve months from date of receipt at -70 ℃ signal transduction pathways as well as immunological control. Originally discovered as an antiviral substance, the efficacy of IFN-alpha in malignant, Predicted N terminal: Glu viral, immunological, angiogenic, inflammatory, and fibrotic diseases suggests a spectrum of interrelated pathophysiologies. -

Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association Between BMI and Adult-Onset Non- Atopic
Supplementary Material DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non- Atopic Asthma Ayoung Jeong 1,2, Medea Imboden 1,2, Akram Ghantous 3, Alexei Novoloaca 3, Anne-Elie Carsin 4,5,6, Manolis Kogevinas 4,5,6, Christian Schindler 1,2, Gianfranco Lovison 7, Zdenko Herceg 3, Cyrille Cuenin 3, Roel Vermeulen 8, Deborah Jarvis 9, André F. S. Amaral 9, Florian Kronenberg 10, Paolo Vineis 11,12 and Nicole Probst-Hensch 1,2,* 1 Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; [email protected] (A.J.); [email protected] (M.I.); [email protected] (C.S.) 2 Department of Public Health, University of Basel, 4001 Basel, Switzerland 3 International Agency for Research on Cancer, 69372 Lyon, France; [email protected] (A.G.); [email protected] (A.N.); [email protected] (Z.H.); [email protected] (C.C.) 4 ISGlobal, Barcelona Institute for Global Health, 08003 Barcelona, Spain; [email protected] (A.-E.C.); [email protected] (M.K.) 5 Universitat Pompeu Fabra (UPF), 08002 Barcelona, Spain 6 CIBER Epidemiología y Salud Pública (CIBERESP), 08005 Barcelona, Spain 7 Department of Economics, Business and Statistics, University of Palermo, 90128 Palermo, Italy; [email protected] 8 Environmental Epidemiology Division, Utrecht University, Institute for Risk Assessment Sciences, 3584CM Utrecht, Netherlands; [email protected] 9 Population Health and Occupational Disease, National Heart and Lung Institute, Imperial College, SW3 6LR London, UK; [email protected] (D.J.); [email protected] (A.F.S.A.) 10 Division of Genetic Epidemiology, Medical University of Innsbruck, 6020 Innsbruck, Austria; [email protected] 11 MRC-PHE Centre for Environment and Health, School of Public Health, Imperial College London, W2 1PG London, UK; [email protected] 12 Italian Institute for Genomic Medicine (IIGM), 10126 Turin, Italy * Correspondence: [email protected]; Tel.: +41-61-284-8378 Int. -
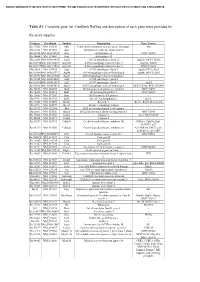
Table S1. Complete Gene List. Genbank Refseq and Description of Each Gene Were Provided By
Document downloaded from http://www.elsevier.es, day 24/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Table S1. Complete gene list. GenBank RefSeq and description of each gene were provided by the array supplier. Unigene GeneBank Symbol Description Gene Name/s Rn.11422 NM_033230 Akt1 V-akt murine thymoma viral oncogene homolog 1 Akt Rn.2104 NM_019288 App Amyloid beta (A4) precursor protein - Rn.23323 NM_001034933 Arsa Arylsulfatase A MGC125207 Rn.94004 NM_033443 Arsb Arylsulfatase B - Rn.6224 NM_001038495 Atg12 ATG12 autophagy related 12 Apg12l, MGC125080 Rn.101734NM_001108809 Atg16l1 ATG16 autophagy related 16-like 1 Apg16l, Wdr30 Rn.104199NM_001191560 Atg16l2 ATG16 autophagy related 16-like 2 RGD1311400 Rn.3084 NM_134394 Atg3 ATG3 autophagy related 3 Apg3l, PIG-1, Pig1 Rn.163086NM_001025711 Atg4b ATG4 autophagy related 4 homolog B Apg4b, MGC112887 Rn.23378 NM_001107948 Atg4c ATG4 autophagy related 4 homolog C - Rn.98385 NM_001014250 Atg5 ATG5 autophagy related 5 - Rn.162765NM_001012097 Atg7 ATG7 autophagy related 7 Apg7l Rn.35248 NM_001014218 Atg9a ATG9 autophagy related 9 homolog A MGC105908, RGD1310450 Rn.36696 NM_022698 Bad BCL2-associated agonist of cell death MGC72439 Rn.14598 NM_053812 Bak1 BCL2-antagonist/killer 1 MGC108627 Rn.10668 NM_017059 Bax Bcl2-associated X protein - Rn.9996 NM_016993 Bcl2 B-cell CLL/lymphoma 2 Bcl-2 Rn.10323 NM_031535 Bcl2l1 Bcl2-like 1 Bcl-xl, Bcl2l, Bclx, bcl-X Rn.2776 NM_053739 Becn1 Beclin 1, autophagy related - Rn.31142 NM_022684 -

Interferonsource.Com What's Inside New Products Research Tools Protocols & Tips Product Citations What's Your IFN-Α
Interferon Matters A quarterly newsletter by PBL InterferonSource interferonsource.com May 2007, Issue 2 u What’s Inside What’s Your IFN-a Type? New Products Are all Type I IFNs created equal? Of Mice and Men page 4 & 5 Type I Interferons were the first cytokines to be The biological significance of the expression of discovered. They are a family of homologous cytokines so many different IFN-a subtypes has yet to be Rhesus/Cynomolgus originally identified for their antiviral activity but have determined. However, reports suggest that they show IFN-a2 Subtype also been shown to have both anti-proliferative and quantitatively distinct anti-viral, anti-proliferative, immunomodulatory activities. In humans and mice and killer cell-stimulatory activities. It is therefore Mouse IFN-a4 Subtype they are encoded by an intronless multigene family of importance for researchers to characterize and clustered on human chromosome 9 and murine study these IFN-a subtypes and their possible post- chromosome 4 respectively. translational modifications. Research Tools Also in both species there are multiple Interferon- The common models for IFN-a subtypes study are page 6 & 7 Alpha (IFN-a) genes and one Interferon-Beta (IFN-b) human and mouse. In 1998, Lawson et al showed that Human IFN-a gene. Other Type I Interferon genes described include there are in vivo differences in the antiviral activity of the Interferon-Omega (IFN-w) gene, murine limitin the mouse IFN-a subtypes.1 Using an experimental Multi-Subtype ELISA Kit gene, as well as the human/murine Interferon-Epilson, mouse model of IFN transgene expression in vivo, TM Interferon-Kappa, and Interferon-Tau (IFN-e, IFN-k, and the authors showed that mouse IFN-a1 has a greater iLite Human IFN-a Kit IFN-t). -

Bufalin Induces G2/M Phase Arrest and Triggers Autophagy Via the TNF, JNK, BECN-1 and ATG8 Pathway in Human Hepatoma Cells
338 INTERNATIONAL JOURNAL OF ONCOLOGY 43: 338-348, 2013 Bufalin induces G2/M phase arrest and triggers autophagy via the TNF, JNK, BECN-1 and ATG8 pathway in human hepatoma cells CHIN-MU HSU1-3, YUHSIN TSAI1,2, LEI WAN1-3* and FUU-JEN TSAI2,4,5* 1Graduate Institute of Chinese Medicine, 2School of Chinese Medicine, China Medical University; Departments of 3Medical Research and 4Medical Genetics, Pediatrics and Medical Research, China Medical University Hospital, 40402 Taichung; 5Department of Biotechnology, Asia University, 41354 Taichung, Taiwan, R.O.C. Received January 16, 2013; Accepted March 18, 2013 DOI: 10.3892/ijo.2013.1942 Abstract. Liver cancer is the fifth most common cause for anti-hepatoma pharmacology for pre-clinical drug investi- of cancer death worldwide. The study of more effective gation. In this case, bufalin was capable of inducing hepatoma anti-hepatoma drugs is urgently required. Bufalin has been cell autophagy, suggesting a potential regimen for single or isolated from a traditional Chinese medicine and possesses combined chemotherapy to overcome hepatoma in clinical less toxicity to normal cells. However, it has been found to practice. inhibit growth of cancer cells. In this study, we aimed to investigate the efficacy and mechanism of bufalin in Huh7, Introduction Hep3B and HA22T human hepatoma cells. The three cell lines were treated with bufalin, the proliferation was detected Liver cancer shows high incident and mortality all over the by WST-1 assay and cell cycle was detected by flow cytom- world. It ranks the fifth and seventh most diagnosed cancer etry analysis. The results showed that bufalin inhibited the worldwide for male and female, respectively (1), and is the proliferation of hepatoma cells and regulated the hepatoma second and sixth leading cause of cancer death for man and cell death program in a dose- and time-dependent manner women, respectively (1). -

Cutting Edge: Endogenous IFN-Β Regulates Survival and Development of Transitional B Cells
Cutting Edge: Endogenous IFN-β Regulates Survival and Development of Transitional B Cells This information is current as Jennie A. Hamilton, Qi Wu, PingAr Yang, Bao Luo, of September 28, 2021. Shanrun Liu, Huixian Hong, Jun Li, Mark R. Walter, Eleanor N. Fish, Hui-Chen Hsu and John D. Mountz J Immunol 2017; 199:2618-2623; Prepublished online 13 September 2017; doi: 10.4049/jimmunol.1700888 Downloaded from http://www.jimmunol.org/content/199/8/2618 Supplementary http://www.jimmunol.org/content/suppl/2017/09/13/jimmunol.170088 Material 8.DCSupplemental http://www.jimmunol.org/ References This article cites 25 articles, 14 of which you can access for free at: http://www.jimmunol.org/content/199/8/2618.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 28, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Th eJournal of Cutting Edge Immunology Cutting Edge: Endogenous IFN-b Regulates Survival and Development of Transitional B Cells Jennie A. -

Type I IFN Is Necessary and Sufficient for Inflammation-Induced Red Blood Cell Alloimmunization in Mice
Type I IFN Is Necessary and Sufficient for Inflammation-Induced Red Blood Cell Alloimmunization in Mice This information is current as David R. Gibb, Jingchun Liu, Prabitha Natarajan, Manjula of September 23, 2021. Santhanakrishnan, David J. Madrid, Stephanie C. Eisenbarth, James C. Zimring, Akiko Iwasaki and Jeanne E. Hendrickson J Immunol 2017; 199:1041-1050; Prepublished online 19 June 2017; Downloaded from doi: 10.4049/jimmunol.1700401 http://www.jimmunol.org/content/199/3/1041 Supplementary http://www.jimmunol.org/content/suppl/2017/06/17/jimmunol.170040 http://www.jimmunol.org/ Material 1.DCSupplemental References This article cites 67 articles, 20 of which you can access for free at: http://www.jimmunol.org/content/199/3/1041.full#ref-list-1 Why The JI? Submit online. by guest on September 23, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Type I IFN Is Necessary and Sufficient for Inflammation-Induced Red Blood Cell Alloimmunization in Mice David R. -

Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium
Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium tuberculosis Infection via STING-Dependent Type 2 Immunity Sulayman Benmerzoug, Badreddine Bounab, Stephanie Rose, David Gosset, Franck Biet, Thierry Cochard, Aurore Xavier, Nathalie Rouxel, Louis Fauconnier, William G.C. Horsnell, et al. To cite this version: Sulayman Benmerzoug, Badreddine Bounab, Stephanie Rose, David Gosset, Franck Biet, et al.. Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium tuberculosis Infection via STING-Dependent Type 2 Immunity. Cell Reports, Elsevier Inc, 2019, 27 (9), pp.2649-2664.e5. 10.1016/j.celrep.2019.04.110. hal-02147129 HAL Id: hal-02147129 https://hal.archives-ouvertes.fr/hal-02147129 Submitted on 26 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution - NonCommercial - NoDerivatives| 4.0 International License Article Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium tuberculosis Infection via STING-Dependent Type 2 Immunity Graphical Abstract Authors Sulayman Benmerzoug, Badreddine Bounab, Ste´ phanie Rose, ..., Bernhard Ryffel, Dieudonnee Togbe, Valerie F.J. Quesniaux Correspondence [email protected] In Brief Benmerzoug et al. mechanistically dissect how sterile lung inflammation induced by silica exacerbates M. tuberculosis infection (silicotuberculosis).