Site-Specific Regulation of Cell Cycle and DNA Repair in Post-Mitotic GABA Cells in Schizophrenic Versus Bipolars
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Investigating the Role of Cdk11in Animal Cytokinesis
Investigating the Role of CDK11 in Animal Cytokinesis by Thomas Clifford Panagiotou A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Molecular Genetics University of Toronto © Copyright by Thomas Clifford Panagiotou (2020) Investigating the Role of CDK11 in Animal Cytokinesis Thomas Clifford Panagiotou Master of Science Department of Molecular Genetics University of Toronto 2020 Abstract Finely tuned spatio-temporal regulation of cell division is required for genome stability. Cytokinesis constitutes the final stages of cell division, from chromosome segregation to the physical separation of cells, abscission. Abscission is tightly regulated to ensure it occurs after earlier cytokinetic events, like the maturation of the stem body, the regulatory platform for abscission. Active Aurora B kinase enforces the abscission checkpoint, which blocks abscission until chromosomes have been cleared from the cytokinetic machinery. Currently, it is unclear how this checkpoint is overcome. Here, I demonstrate that the cyclin-dependent kinase CDK11 is required for cytokinesis. Both inhibition and depletion of CDK11 block abscission. Furthermore, the mitosis-specific CDK11p58 kinase localizes to the stem body, where its kinase activity rescues the defects of CDK11 depletion and inhibition. These results suggest a model whereby CDK11p58 antagonizes Aurora B kinase to overcome the abscission checkpoint to allow for successful completion of cytokinesis. ii Acknowledgments I am very grateful for the support of my family and friends throughout my studies. I would also like to express my deep gratitude to Wilde Lab members, both past and present, for their advice and collaboration. In particular, I am very grateful to Matthew Renshaw, whose work comprises part of this thesis. -

Support Info
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2014 Supporting Information Design and synthesis of pyrrole–5-(2,6-dichlorobenzyl)sulfonylindolin-2-ones with C- 3’ side chains as potent Met kinase inhibitors Chia-Wei Liu,a Chun-Liang Lai,a Yu-Hsiang Lin,a Li-Wei Teng,a Sheng-chuan Yang,a Win-Yin Wei,a Shu Fu Lin,a Ju-Ying Yang,a Hung-Jyun Huang,a Ru-Wen Wang,a Chao-Cheng Chiang,a Mei-Hui Lee,a Yu- Chuan Wang,b Shih-Hsien Chuang,a Jia-Ming Chang,a Ying-Shuan E. Lee,a and Jiann-Jyh Huang*a,b aDevelopment Center for Biotechnology, No. 101, Lane 169, Kangning St., Xizhi District, New Taipei City 22180, Taiwan bDepartment of Applied Chemistry, National Chiayi University, No. 300, Syuefu Rd., Chiayi City 60004, Taiwan *Corresponding Author. Tel.: +886 5 271 7959; Fax: +886 5 271 7901. E-mail address: [email protected] (J.-J. Huang) Table of Contents: Page Supporting Figure. Ligplot diagrams of the ATP binding site of Met S2 complexed with compounds 2 and 20. Supporting Table. Kinase profiling data of compound 20. S3 References S10 - S1 - Supporting Figure. Ligplot diagrams1 of the ATP binding site of Met complexed with compounds 2 and 20: (A) Met with 2, and (B) Met with 20. - S2 - Supporting Table. Kinase profiling data of 20. Ambit KinomeScan Kinase Profiling (1.0 μM test concentration): Percentage of Percentage of Ambit Gene Symbol control (%) Ambit Gene Symbol control (%) 20 20 AAK1 68 ARK5 27 ABL1(E255K)-phosphorylated 85 ASK1 100 ABL1(F317I)-nonphosphorylated 78 ASK2 67 -

Profiling Data
Compound Name DiscoveRx Gene Symbol Entrez Gene Percent Compound Symbol Control Concentration (nM) JNK-IN-8 AAK1 AAK1 69 1000 JNK-IN-8 ABL1(E255K)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317I)-nonphosphorylated ABL1 87 1000 JNK-IN-8 ABL1(F317I)-phosphorylated ABL1 100 1000 JNK-IN-8 ABL1(F317L)-nonphosphorylated ABL1 65 1000 JNK-IN-8 ABL1(F317L)-phosphorylated ABL1 61 1000 JNK-IN-8 ABL1(H396P)-nonphosphorylated ABL1 42 1000 JNK-IN-8 ABL1(H396P)-phosphorylated ABL1 60 1000 JNK-IN-8 ABL1(M351T)-phosphorylated ABL1 81 1000 JNK-IN-8 ABL1(Q252H)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(Q252H)-phosphorylated ABL1 56 1000 JNK-IN-8 ABL1(T315I)-nonphosphorylated ABL1 100 1000 JNK-IN-8 ABL1(T315I)-phosphorylated ABL1 92 1000 JNK-IN-8 ABL1(Y253F)-phosphorylated ABL1 71 1000 JNK-IN-8 ABL1-nonphosphorylated ABL1 97 1000 JNK-IN-8 ABL1-phosphorylated ABL1 100 1000 JNK-IN-8 ABL2 ABL2 97 1000 JNK-IN-8 ACVR1 ACVR1 100 1000 JNK-IN-8 ACVR1B ACVR1B 88 1000 JNK-IN-8 ACVR2A ACVR2A 100 1000 JNK-IN-8 ACVR2B ACVR2B 100 1000 JNK-IN-8 ACVRL1 ACVRL1 96 1000 JNK-IN-8 ADCK3 CABC1 100 1000 JNK-IN-8 ADCK4 ADCK4 93 1000 JNK-IN-8 AKT1 AKT1 100 1000 JNK-IN-8 AKT2 AKT2 100 1000 JNK-IN-8 AKT3 AKT3 100 1000 JNK-IN-8 ALK ALK 85 1000 JNK-IN-8 AMPK-alpha1 PRKAA1 100 1000 JNK-IN-8 AMPK-alpha2 PRKAA2 84 1000 JNK-IN-8 ANKK1 ANKK1 75 1000 JNK-IN-8 ARK5 NUAK1 100 1000 JNK-IN-8 ASK1 MAP3K5 100 1000 JNK-IN-8 ASK2 MAP3K6 93 1000 JNK-IN-8 AURKA AURKA 100 1000 JNK-IN-8 AURKA AURKA 84 1000 JNK-IN-8 AURKB AURKB 83 1000 JNK-IN-8 AURKB AURKB 96 1000 JNK-IN-8 AURKC AURKC 95 1000 JNK-IN-8 -
Supplementary Table 2 Supplementary Table 1
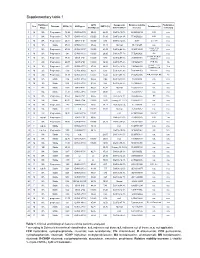
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Genome-Wide Sirna Screen for Modulators of Cell Death Induced by Proteasome Inhibitor Bortezomib
Published OnlineFirst May 11, 2010; DOI: 10.1158/0008-5472.CAN-09-4428 Published OnlineFirst on May 11, 2010 as 10.1158/0008-5472.CAN-09-4428 Integrated Systems and Technologies Cancer Research Genome-Wide siRNA Screen for Modulators of Cell Death Induced by Proteasome Inhibitor Bortezomib Siquan Chen1, Jonathan L. Blank2, Theodore Peters1, Xiaozhen J. Liu2, David M. Rappoli1, Michael D. Pickard3, Saurabh Menon1, Jie Yu2, Denise L. Driscoll2, Trupti Lingaraj2, Anne L. Burkhardt2, Wei Chen2, Khristofer Garcia1, Darshan S. Sappal2, Jesse Gray1, Paul Hales1, Patrick J. Leroy2, John Ringeling1, Claudia Rabino2, James J. Spelman2, Jay P. Morgenstern1, and Eric S. Lightcap2 Abstract Multiple pathways have been proposed to explain how proteasome inhibition induces cell death, but me- chanisms remain unclear. To approach this issue, we performed a genome-wide siRNA screen to evaluate the genetic determinants that confer sensitivity to bortezomib (Velcade (R); PS-341). This screen identified 100 genes whose knockdown affected lethality to bortezomib and to a structurally diverse set of other proteasome inhibitors. A comparison of three cell lines revealed that 39 of 100 genes were commonly linked to cell death. We causally linked bortezomib-induced cell death to the accumulation of ASF1B, Myc, ODC1, Noxa, BNIP3, Gadd45α, p-SMC1A, SREBF1, and p53. Our results suggest that proteasome inhibition promotes cell death primarily by dysregulating Myc and polyamines, interfering with protein translation, and disrupting essential DNA damage repair pathways, leading to programmed cell death. Cancer Res; 70(11); OF1–9. ©2010 AACR. Introduction active oxygen species (ROS), stabilization of Myc and Noxa, and induction of endoplasmic reticulum (ER) stress, any of The primary targets of most cancer chemotherapies are which may trigger cell death (1–4). -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

Study on Apoptosis in Human Deciduous Tooth Pulp Cells Original
Yuko Okai et al.: Apoptosis in Human Deciduous Tooth Pulp Cells Journal of Hard Tissue Biology 21[4] (2012) p413-420 © 2012 The Hard Tissue Biology Network Association Printed in Japan, All rights reserved. CODEN-JHTBFF, ISSN 1341-7649 Original Study on Apoptosis in Human Deciduous Tooth Pulp Cells Yuko Okai1), Kyoko Harada1), Kiyoshi Ohura2) and Kenji Arita1) 1)Department of Pediatric Dentistry, Osaka Dental University,Osaka, Japan 2)Department of Pharmacology, Osaka Dental University, Osaka, Japan (Accepted for publication, July 5, 2012) Abstract: Dental pulp plays an important role in tooth vitality. The pulp of deciduous tooth thus shows vigorous metabolism and repeated, vigorous cell regeneration and proliferation. Furthermore, natural, programmed cell death (apoptosis) frequently occurs as part of this cell regeneration and proliferation process. Apoptosis is defined as cell death governed by intracellular signals (active death of the cell triggered by changes in physiological or external conditions), and the birth of new cells occurs simultaneously with cell death. Apoptosis thus functions as a driving force for morphological and other changes in organisms, and is essential for phenomena such as ontogenetic processes or aging. However, In order to clarify the mechanism of apoptosis in the deciduous tooth pulp cells, we used SuperArray analysis and immunostaining to examine both deciduous and permanent tooth pulp cells. Of the 84 major genes involved in apoptosis, seven showed greater expression in the deciduous tooth pulp cells than in the permanent tooth pulp cells. Gene expression of TP53BP2, CRADD, BAK1, BIRC3, CASP8, CASP2 and TP73 in the deciduous tooth pulp were 16.1, 9.0, 8.5, 8.1, 5.9, 5.8 and 3.2 times greater than those in the permanent tooth pulp cells, respectively. -

Hsa-Mir-21-5P
Supporting Information An exosomal urinary miRNA signature for early diagnosis of renal fibrosis in Lupus Nephritis 1 2 2 1 1 Cristina Solé , Teresa Moliné , Marta Vidal , Josep Ordi-Ros , Josefina Cortés-Hernández 1Hospital Universitari Vall d’Hebron, Vall d’Hebron Research Institute (VHIR), Lupus Unit, Barcelona, Spain; [email protected] (C.S); [email protected] (J.O-R.); [email protected] (J.C-H.) 2 Hospital Universitari Vall d’Hebron, Department of Renal Pathology, Barcelona, Spain; [email protected] (T.M); [email protected] (M.V). INDEX 1. SI Materials and Methods......................................................................................3 2. SI References…………………………………………………………………………….7 2. SI Figures................................................................................................................8 4. SI Tables................................................................................................................21 2 SI Materials and Methods Patients and samples Patients with biopsy-proven active LN were recruited from the Lupus Unit at Vall d’Hebron Hospital (N=45). All patients fulfilled at least 4 of the American College of rheumatology (ACR) revised classification criteria for SLE [1]. Healthy donors were used as controls (N=20). Urine samples were collected from each patient 1 day before renal biopsy and processed immediately to be stored at -80ºC. Patients with urinary tract infection, diabetes mellitus, pregnancy, malignancy and non-lupus-related renal failure were excluded. In addition, key laboratory measurements were obtained including complement levels (C3 and C4), anti-double-stranded DNA (anti-dsDNA), 24-h proteinuria, blood urea nitrogen (BUN), serum creatinine and the estimated glomerular filtration ratio (eGFR) using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula [2]. SLE disease activity was assessed by the SLE Disease Activity Index 2000 update (SLEDAI-2Ks; range 0–105) [3]. -

The Emerging Roles and Therapeutic Potential of Cyclin- Dependent Kinase 11 (CDK11) in Human Cancer
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 26 Review The emerging roles and therapeutic potential of cyclin- dependent kinase 11 (CDK11) in human cancer Yubing Zhou1,2, Jacson K. Shen2, Francis J. Hornicek2, Quancheng Kan1 and Zhenfeng Duan1,2 1 Department of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan, People’s Republic of China 2 Sarcoma Biology Laboratory, Center for Sarcoma and Connective Tissue Oncology, Massachusetts General Hospital, Boston, MA, United States of America Correspondence to: Quancheng Kan, email: [email protected] Correspondence to: Zhenfeng Duan, email: [email protected] Keywords: CDK11, CDKs inhibitor, cell cycle, therapeutic target, cancer therapy Received: January 20, 2016 Accepted: March 28, 2016 Published: March 31, 2016 ABSTRACT Overexpression and/or hyperactivation of cyclin-dependent kinases (CDKs) are common features of most cancer types. CDKs have been shown to play important roles in tumor cell proliferation and growth by controlling cell cycle, transcription, and RNA splicing. CDK4/6 inhibitor palbociclib has been recently approved by the FDA for the treatment of breast cancer. CDK11 is a serine/threonine protein kinase in the CDK family and recent studies have shown that CDK11 also plays critical roles in cancer cell growth and proliferation. A variety of genetic and epigenetic events may cause universal overexpression of CDK11 in human cancers. Inhibition of CDK11 has been shown to lead to cancer cell death and apoptosis. Significant evidence has suggested that CDK11 may be a novel and promising therapeutic target for the treatment of cancers. This review will focus on the emerging roles of CDK11 in human cancers, and provide a proof-of-principle for continued efforts toward targeting CDK11 for effective cancer treatment. -

Identification of Differentially Expressed Genes in Human Bladder Cancer Through Genome-Wide Gene Expression Profiling
521-531 24/7/06 18:28 Page 521 ONCOLOGY REPORTS 16: 521-531, 2006 521 Identification of differentially expressed genes in human bladder cancer through genome-wide gene expression profiling KAZUMORI KAWAKAMI1,3, HIDEKI ENOKIDA1, TOKUSHI TACHIWADA1, TAKENARI GOTANDA1, KENGO TSUNEYOSHI1, HIROYUKI KUBO1, KENRYU NISHIYAMA1, MASAKI TAKIGUCHI2, MASAYUKI NAKAGAWA1 and NAOHIKO SEKI3 1Department of Urology, Graduate School of Medical and Dental Sciences, Kagoshima University, 8-35-1 Sakuragaoka, Kagoshima 890-8520; Departments of 2Biochemistry and Genetics, and 3Functional Genomics, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received February 15, 2006; Accepted April 27, 2006 Abstract. Large-scale gene expression profiling is an effective CKS2 gene not only as a potential biomarker for diagnosing, strategy for understanding the progression of bladder cancer but also for staging human BC. This is the first report (BC). The aim of this study was to identify genes that are demonstrating that CKS2 expression is strongly correlated expressed differently in the course of BC progression and to with the progression of human BC. establish new biomarkers for BC. Specimens from 21 patients with pathologically confirmed superficial (n=10) or Introduction invasive (n=11) BC and 4 normal bladder samples were studied; samples from 14 of the 21 BC samples were subjected Bladder cancer (BC) is among the 5 most common to microarray analysis. The validity of the microarray results malignancies worldwide, and the 2nd most common tumor of was verified by real-time RT-PCR. Of the 136 up-regulated the genitourinary tract and the 2nd most common cause of genes we detected, 21 were present in all 14 BCs examined death in patients with cancer of the urinary tract (1-7). -

Modes of Cell Death Induced by Photodynamic Therapy Using Zinc Phthalocyanine in Lung Cancer Cells Grown As a Monolayer and Three-Dimensional Multicellular Spheroids
molecules Article Modes of Cell Death Induced by Photodynamic Therapy Using Zinc Phthalocyanine in Lung Cancer Cells Grown as a Monolayer and Three-Dimensional Multicellular Spheroids Sello L. Manoto, Nicolette Houreld, Natasha Hodgkinson and Heidi Abrahamse * Laser Research Centre, Faculty of Health Sciences, University of Johannesburg, P.O. Box 17011, Doornfontein 2028, South Africa; [email protected] (S.L.M.); [email protected] (N.H.); [email protected] (N.H.) * Correspondence: [email protected]; Tel.: +27-11-559-6406; Fax: +27-11-559-6884 Academic Editors: Augusto C. Tomé, João Paulo C. Tomé and Michael Hanack Received: 8 March 2017; Accepted: 9 May 2017; Published: 16 May 2017 Abstract: Photodynamic therapy (PDT) involves interaction of a photosensitizer, light, and molecular oxygen which produces singlet oxygen and subsequent tumour eradication. The development of second generation photosensitizers, such as phthalocyanines, has improved this technology. Customary monolayer cell culture techniques are, unfortunately, too simple to replicate treatment effects in vivo. Multicellular tumour spheroids may provide a better alternative since they mimic aspects of the human tumour environment. This study aimed to profile 84 genes involved in apoptosis following treatment with PDT on lung cancer cells (A549) grown in a monolayer versus three-dimensional multicellular tumour spheroids (250 and 500 µm). Gene expression profiling 2 was performed 24 h post irradiation (680 nm; 5 J/cm ) with zinc sulfophthalocyanine (ZnPcSmix) to determine the genes involved in apoptotic cell death. In the monolayer cells, eight pro-apoptotic genes were upregulated, and two were downregulated. In the multicellular tumour spheroids (250 µm) there was upregulation of only 1 gene while there was downregulation of 56 genes. -

A Four-Gene Decision Tree Signature
Published OnlineFirst October 10, 2018; DOI: 10.1158/1535-7163.MCT-18-0243 Companion Diagnostic, Pharmacogenomic, and Cancer Biomarkers Molecular Cancer Therapeutics A Four-gene Decision Tree Signature Classification of Triple-negative Breast Cancer: Implications for Targeted Therapeutics Jelmar Quist1,2,3, Hasan Mirza1,2,3, Maggie C.U. Cheang4,5, Melinda L. Telli6, Joyce A. O'Shaughnessy7, Christopher J. Lord5,8, Andrew N.J. Tutt2,3,5, and Anita Grigoriadis1,2,3 Abstract The molecular complexity of triple-negative breast cancers in early diagnosed TNBCs. In TNBC patients with metastatic (TNBCs) provides a challenge for patient management. We set disease, the MC6 proportion was reduced to 25%, and was out to characterize this heterogeneous disease by combining independently associated with a higher response rate to transcriptomics and genomics data, with the aim of revealing platinum-based chemotherapy. In TNBC cell line data, convergent pathway dependencies with the potential for treat- platinum sensitivity was recapitulated, and a sensitivity ment intervention. A Bayesian algorithm was used to integrate to the inhibition of the phosphatase PPM1D was revealed. molecular profiles in two TNBC cohorts, followed by valida- Molecularly, MC6-TNBCs displayed high levels of telomeric þ þ tion using five independent cohorts (n ¼ 1,168), including allelic imbalances, enrichment of CD4 and CD8 immune three clinical trials. A four-gene decision tree signature was signatures, and reduced expression of genes negatively reg- identified, which robustly classified TNBCs into six subtypes. ulating the MAPK signaling pathway. These observations All four genes in the signature (EXO1, TP53BP2, FOXM1, and suggest that our integrative classification approach may RSU1) are associated with either genomic instability, malig- identify TNBC patients with discernible and theoretically nant growth, or treatment response.