Hsa-Mir-21-5P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
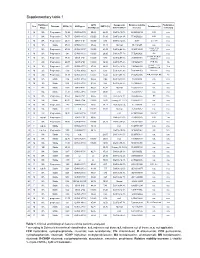
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

Study on Apoptosis in Human Deciduous Tooth Pulp Cells Original
Yuko Okai et al.: Apoptosis in Human Deciduous Tooth Pulp Cells Journal of Hard Tissue Biology 21[4] (2012) p413-420 © 2012 The Hard Tissue Biology Network Association Printed in Japan, All rights reserved. CODEN-JHTBFF, ISSN 1341-7649 Original Study on Apoptosis in Human Deciduous Tooth Pulp Cells Yuko Okai1), Kyoko Harada1), Kiyoshi Ohura2) and Kenji Arita1) 1)Department of Pediatric Dentistry, Osaka Dental University,Osaka, Japan 2)Department of Pharmacology, Osaka Dental University, Osaka, Japan (Accepted for publication, July 5, 2012) Abstract: Dental pulp plays an important role in tooth vitality. The pulp of deciduous tooth thus shows vigorous metabolism and repeated, vigorous cell regeneration and proliferation. Furthermore, natural, programmed cell death (apoptosis) frequently occurs as part of this cell regeneration and proliferation process. Apoptosis is defined as cell death governed by intracellular signals (active death of the cell triggered by changes in physiological or external conditions), and the birth of new cells occurs simultaneously with cell death. Apoptosis thus functions as a driving force for morphological and other changes in organisms, and is essential for phenomena such as ontogenetic processes or aging. However, In order to clarify the mechanism of apoptosis in the deciduous tooth pulp cells, we used SuperArray analysis and immunostaining to examine both deciduous and permanent tooth pulp cells. Of the 84 major genes involved in apoptosis, seven showed greater expression in the deciduous tooth pulp cells than in the permanent tooth pulp cells. Gene expression of TP53BP2, CRADD, BAK1, BIRC3, CASP8, CASP2 and TP73 in the deciduous tooth pulp were 16.1, 9.0, 8.5, 8.1, 5.9, 5.8 and 3.2 times greater than those in the permanent tooth pulp cells, respectively. -

Modes of Cell Death Induced by Photodynamic Therapy Using Zinc Phthalocyanine in Lung Cancer Cells Grown As a Monolayer and Three-Dimensional Multicellular Spheroids
molecules Article Modes of Cell Death Induced by Photodynamic Therapy Using Zinc Phthalocyanine in Lung Cancer Cells Grown as a Monolayer and Three-Dimensional Multicellular Spheroids Sello L. Manoto, Nicolette Houreld, Natasha Hodgkinson and Heidi Abrahamse * Laser Research Centre, Faculty of Health Sciences, University of Johannesburg, P.O. Box 17011, Doornfontein 2028, South Africa; [email protected] (S.L.M.); [email protected] (N.H.); [email protected] (N.H.) * Correspondence: [email protected]; Tel.: +27-11-559-6406; Fax: +27-11-559-6884 Academic Editors: Augusto C. Tomé, João Paulo C. Tomé and Michael Hanack Received: 8 March 2017; Accepted: 9 May 2017; Published: 16 May 2017 Abstract: Photodynamic therapy (PDT) involves interaction of a photosensitizer, light, and molecular oxygen which produces singlet oxygen and subsequent tumour eradication. The development of second generation photosensitizers, such as phthalocyanines, has improved this technology. Customary monolayer cell culture techniques are, unfortunately, too simple to replicate treatment effects in vivo. Multicellular tumour spheroids may provide a better alternative since they mimic aspects of the human tumour environment. This study aimed to profile 84 genes involved in apoptosis following treatment with PDT on lung cancer cells (A549) grown in a monolayer versus three-dimensional multicellular tumour spheroids (250 and 500 µm). Gene expression profiling 2 was performed 24 h post irradiation (680 nm; 5 J/cm ) with zinc sulfophthalocyanine (ZnPcSmix) to determine the genes involved in apoptotic cell death. In the monolayer cells, eight pro-apoptotic genes were upregulated, and two were downregulated. In the multicellular tumour spheroids (250 µm) there was upregulation of only 1 gene while there was downregulation of 56 genes. -

A Four-Gene Decision Tree Signature
Published OnlineFirst October 10, 2018; DOI: 10.1158/1535-7163.MCT-18-0243 Companion Diagnostic, Pharmacogenomic, and Cancer Biomarkers Molecular Cancer Therapeutics A Four-gene Decision Tree Signature Classification of Triple-negative Breast Cancer: Implications for Targeted Therapeutics Jelmar Quist1,2,3, Hasan Mirza1,2,3, Maggie C.U. Cheang4,5, Melinda L. Telli6, Joyce A. O'Shaughnessy7, Christopher J. Lord5,8, Andrew N.J. Tutt2,3,5, and Anita Grigoriadis1,2,3 Abstract The molecular complexity of triple-negative breast cancers in early diagnosed TNBCs. In TNBC patients with metastatic (TNBCs) provides a challenge for patient management. We set disease, the MC6 proportion was reduced to 25%, and was out to characterize this heterogeneous disease by combining independently associated with a higher response rate to transcriptomics and genomics data, with the aim of revealing platinum-based chemotherapy. In TNBC cell line data, convergent pathway dependencies with the potential for treat- platinum sensitivity was recapitulated, and a sensitivity ment intervention. A Bayesian algorithm was used to integrate to the inhibition of the phosphatase PPM1D was revealed. molecular profiles in two TNBC cohorts, followed by valida- Molecularly, MC6-TNBCs displayed high levels of telomeric þ þ tion using five independent cohorts (n ¼ 1,168), including allelic imbalances, enrichment of CD4 and CD8 immune three clinical trials. A four-gene decision tree signature was signatures, and reduced expression of genes negatively reg- identified, which robustly classified TNBCs into six subtypes. ulating the MAPK signaling pathway. These observations All four genes in the signature (EXO1, TP53BP2, FOXM1, and suggest that our integrative classification approach may RSU1) are associated with either genomic instability, malig- identify TNBC patients with discernible and theoretically nant growth, or treatment response. -

Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution
viruses Article Combinatorial Avidity Selection of Mosaic Landscape Phages Targeted at Breast Cancer Cells—An Alternative Mechanism of Directed Molecular Evolution Valery A. Petrenko 1,* , James W. Gillespie 1 , Hai Xu 1,2, Tiffany O’Dell 1 and Laura M. De Plano 1,3 1 Department of Pathobiology, College of Veterinary Medicine, Auburn University, Auburn, AL 36849, USA 2 National Veterinary Biological Medicine Engineering Research Center, Jiangsu Academy of Agricultural Sciences, Nanjing 210014, Jiangsu, China 3 Department of Chemical Sciences, Biological, Pharmaceutical and Environmental, University of Messina, Viale F. Stagno d’Alcontres 31, 98166 Messina, Italy * Correspondence: [email protected]; Tel.: +1-334-844-2897 Received: 2 August 2019; Accepted: 22 August 2019; Published: 26 August 2019 Abstract: Low performance of actively targeted nanomedicines required revision of the traditional drug targeting paradigm and stimulated the development of novel phage-programmed, self-navigating drug delivery vehicles. In the proposed smart vehicles, targeting peptides, selected from phage libraries using traditional principles of affinity selection, are substituted for phage proteins discovered through combinatorial avidity selection. Here, we substantiate the potential of combinatorial avidity selection using landscape phage in the discovery of Short Linear Motifs (SLiMs) and their partner domains. We proved an algorithm for analysis of phage populations evolved through multistage screening of landscape phage libraries against the MDA-MB-231 breast cancer cell line. The suggested combinatorial avidity selection model proposes a multistage accumulation of Elementary Binding Units (EBU), or Core Motifs (CorMs), in landscape phage fusion peptides, serving as evolutionary initiators for formation of SLiMs. Combinatorial selection has the potential to harness directed molecular evolution to create novel smart materials with diverse novel, emergent properties. -

Exploring Extracellular Vesicles Biogenesis in Hypothalamic Cells Through a Heavy Isotope Pulse/Trace Proteomic Approach
cells Article Exploring Extracellular Vesicles Biogenesis in Hypothalamic Cells through a Heavy Isotope Pulse/Trace Proteomic Approach Chee Fan Tan 1,2 , Hui San Teo 2, Jung Eun Park 2, Bamaprasad Dutta 2, Shun Wilford Tse 2, Melvin Khee-Shing Leow 3,4,5 , Walter Wahli 3,6 and Siu Kwan Sze 2,* 1 NTU Institute for Health Technologies, Interdisciplinary Graduate School, Nanyang Technological University, Singapore 637335, Singapore; [email protected] 2 School of Biological Sciences, Nanyang Technological University, Singapore 637551, Singapore; [email protected] (H.S.T.); [email protected] (J.E.P.); [email protected] (B.D.); [email protected] (S.W.T.) 3 Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore 636921, Singapore; [email protected] (M.K.-S.L.); [email protected] (W.W.) 4 Department of Endocrinology, Tan Tock Seng Hospital, Singapore 308433, Singapore 5 Cardiovascular and Metabolic Disorder Program, Duke-NUS Medical School, Singapore 169857, Singapore 6 Center for Integrative Genomics, University of Lausanne, Le Génopode, CH-1015 Lausanne, Switzerland * Correspondence: [email protected]; Tel.: +65-6514-1006; Fax: +65-6791-3856 Received: 24 March 2020; Accepted: 21 May 2020; Published: 25 May 2020 Abstract: Studies have shown that the process of extracellular vesicles (EVs) secretion and lysosome status are linked. When the lysosome is under stress, the cells would secrete more EVs to maintain cellular homeostasis. However, the process that governs lysosomal activity and EVs secretion remains poorly defined and we postulated that certain proteins essential for EVs biogenesis are constantly synthesized and preferentially sorted to the EVs rather than the lysosome. -

Rna-Sequencing Applications: Gene Expression Quantification and Methylator Phenotype Identification
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 8-2013 RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION Guoshuai Cai Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Computational Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Cai, Guoshuai, "RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 386. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/386 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION -

Gene Section Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Review TP53BP2 (tumor protein p53 binding protein, 2) Kathryn Van Hook, Zhiping Wang, Charles Lopez Department of Medicine, Division of Hematology and Medical Oncology, Oregon Health and Science University, Portland, OR, USA (KV, ZW, CL) Published in Atlas Database: January 2011 Online updated version : http://AtlasGeneticsOncology.org/Genes/TP53BP2ID42667ch1q42.html DOI: 10.4267/2042/46004 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2011 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Pseudogene Not known. Other names: 53BP2; ASPP2; BBP; P53BP2; PPP1R13A Protein HGNC (Hugo): TP53BP2 Location: 1q41 Description ASPP2 is a pro-apoptotic protein with a predicted size DNA/RNA of approximately 135 kDa. It is the founding member of a family of ASPP proteins that all share the common Description motifs of four Ankyrin-repeats, a Src-homology 3 The TP53BP2 gene spans about 66 kb on chromosome (SH3) domain, and a Polyproline domain in their C- 1q42.1 on the minus strand (Yang et al., 1997). There terminus (Iwabuchi et al., 1994). The N-terminus of are two transcripts as a result of alternative splicing ASPP2 is thought to be important for regulating its (Takahashi et al., 2004). The transcript variant 1, which apoptotic function and contains a putative Ras- is shorter (4670 bp), does not contain exon 3 and gives association domain as well as a ubiquitin-like fold rise to a longer form of the protein named TP53BPL (Tidow et al., 2007). -

The Role of Tsg101 in the Development of Physiological Cardiac Hypertrophy
The Role of Tsg101 in the Development of Physiological Cardiac Hypertrophy and Cardio-Protection from Endotoxin-Induced Cardiac Dysfunction A dissertation to be submitted to the Graduate School of the University of Cincinnati in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Department of Pharmacology and Systems Physiology, College of Medicine By: Kobina Q. Essandoh B.A. in Biochemistry from Cornell College, 2011 Advisor and Committee Chair: Guo-Chang Fan, Ph.D. Abstract In this dissertation, the functional role of Tumor susceptibility gene (Tsg101) in the regulation of physiological cardiac hypertrophy and endotoxin-induced cardiac dysfunction was explored. Development of physiological cardiac hypertrophy has primarily been ascribed to the insulin-like growth factor 1 (IGF-1) and its receptor, IGF-1R, and subsequent activation of the Akt pathway. However, regulation of endosome-mediated recycling and degradation of IGF-1R during physiological hypertrophy has not been investigated. Furthermore, cardiac mitochondrial damage and subsequent inflammation are hallmarks of endotoxin-induced myocardial depression. Activation of the Parkin/PINK1 pathway has been shown to promote autophagy of damaged mitochondria (mitophagy) and protect from endotoxin-induced cardiac dysfunction. Tsg101 has been demonstrated to play diverse roles in the cell including virus budding, cytokinesis, transcriptional regulation, endosomal recycling of receptors and activation of autophagic flux. Hence, the first goal of this dissertation was to elucidate the role of Tg101 in endosome-mediated recycling of IGF-1R in physiological cardiac remodeling. The second goal of this dissertation was to investigate whether Tsg101 regulates mitophagy and thus contribute to endotoxin-caused myocardial dysfunction. -

A Graph-Theoretic Approach to Model Genomic Data and Identify Biological Modules Asscociated with Cancer Outcomes
A Graph-Theoretic Approach to Model Genomic Data and Identify Biological Modules Asscociated with Cancer Outcomes Deanna Petrochilos A dissertation presented in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2013 Reading Committee: Neil Abernethy, Chair John Gennari, Ali Shojaie Program Authorized to Offer Degree: Biomedical Informatics and Health Education UMI Number: 3588836 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMI 3588836 Published by ProQuest LLC (2013). Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code ProQuest LLC. 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, MI 48106 - 1346 ©Copyright 2013 Deanna Petrochilos University of Washington Abstract Using Graph-Based Methods to Integrate and Analyze Cancer Genomic Data Deanna Petrochilos Chair of the Supervisory Committee: Assistant Professor Neil Abernethy Biomedical Informatics and Health Education Studies of the genetic basis of complex disease present statistical and methodological challenges in the discovery of reliable and high-confidence genes that reveal biological phenomena underlying the etiology of disease or gene signatures prognostic of disease outcomes. This dissertation examines the capacity of graph-theoretical methods to model and analyze genomic information and thus facilitate using prior knowledge to create a more discrete and functionally relevant feature space. -

Comparative Analysis of the Ubiquitin-Proteasome System in Homo Sapiens and Saccharomyces Cerevisiae
Comparative Analysis of the Ubiquitin-proteasome system in Homo sapiens and Saccharomyces cerevisiae Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln vorgelegt von Hartmut Scheel aus Rheinbach Köln, 2005 Berichterstatter: Prof. Dr. R. Jürgen Dohmen Prof. Dr. Thomas Langer Dr. Kay Hofmann Tag der mündlichen Prüfung: 18.07.2005 Zusammenfassung I Zusammenfassung Das Ubiquitin-Proteasom System (UPS) stellt den wichtigsten Abbauweg für intrazelluläre Proteine in eukaryotischen Zellen dar. Das abzubauende Protein wird zunächst über eine Enzym-Kaskade mit einer kovalent gebundenen Ubiquitinkette markiert. Anschließend wird das konjugierte Substrat vom Proteasom erkannt und proteolytisch gespalten. Ubiquitin besitzt eine Reihe von Homologen, die ebenfalls posttranslational an Proteine gekoppelt werden können, wie z.B. SUMO und NEDD8. Die hierbei verwendeten Aktivierungs- und Konjugations-Kaskaden sind vollständig analog zu der des Ubiquitin- Systems. Es ist charakteristisch für das UPS, daß sich die Vielzahl der daran beteiligten Proteine aus nur wenigen Proteinfamilien rekrutiert, die durch gemeinsame, funktionale Homologiedomänen gekennzeichnet sind. Einige dieser funktionalen Domänen sind auch in den Modifikations-Systemen der Ubiquitin-Homologen zu finden, jedoch verfügen diese Systeme zusätzlich über spezifische Domänentypen. Homologiedomänen lassen sich als mathematische Modelle in Form von Domänen- deskriptoren (Profile) beschreiben. Diese Deskriptoren können wiederum dazu verwendet werden, mit Hilfe geeigneter Verfahren eine gegebene Proteinsequenz auf das Vorliegen von entsprechenden Homologiedomänen zu untersuchen. Da die im UPS involvierten Homologie- domänen fast ausschließlich auf dieses System und seine Analoga beschränkt sind, können domänen-spezifische Profile zur Katalogisierung der UPS-relevanten Proteine einer Spezies verwendet werden. Auf dieser Basis können dann die entsprechenden UPS-Repertoires verschiedener Spezies miteinander verglichen werden.