Bioinformatics Mining of the Dark Matter Proteome For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Astrin-SKAP Complex Reconstitution Reveals Its Kinetochore
RESEARCH ARTICLE Astrin-SKAP complex reconstitution reveals its kinetochore interaction with microtubule-bound Ndc80 David M Kern1,2, Julie K Monda1,2†, Kuan-Chung Su1†, Elizabeth M Wilson-Kubalek3, Iain M Cheeseman1,2* 1Whitehead Institute for Biomedical Research, Cambridge, United States; 2Department of Biology, Massachusetts Institute of Technology, Cambridge, United States; 3Department of Cell Biology, The Scripps Research Institute, La Jolla, United States Abstract Chromosome segregation requires robust interactions between the macromolecular kinetochore structure and dynamic microtubule polymers. A key outstanding question is how kinetochore-microtubule attachments are modulated to ensure that bi-oriented attachments are selectively stabilized and maintained. The Astrin-SKAP complex localizes preferentially to properly bi-oriented sister kinetochores, representing the final outer kinetochore component recruited prior to anaphase onset. Here, we reconstitute the 4-subunit Astrin-SKAP complex, including a novel MYCBP subunit. Our work demonstrates that the Astrin-SKAP complex contains separable kinetochore localization and microtubule binding domains. In addition, through cross-linking analysis in human cells and biochemical reconstitution, we show that the Astrin-SKAP complex binds synergistically to microtubules with the Ndc80 complex to form an integrated interface. We propose a model in which the Astrin-SKAP complex acts together with the Ndc80 complex to stabilize correctly formed kinetochore-microtubule interactions. *For correspondence: DOI: https://doi.org/10.7554/eLife.26866.001 [email protected] †These authors contributed equally to this work Introduction Competing interests: The The macromolecular kinetochore complex links chromosomes to dynamic microtubule polymers and authors declare that no harnesses the forces generated by microtubule growth and depolymerization to facilitate accurate competing interests exist. -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Chemical Genomics 33
Curr. Issues Mol. Biol. (2002) 4: 33-43. Chemical Genomics 33 Chemical Genomics: A Systematic Approach in Biological Research and Drug Discovery X.F. Steven Zheng1* and Ting-Fung Chan2 synthesis (Russell and Eggleston 2000) and new screening technologies such as small chemical compound (MacBeath 1Department of Pathology and Immunology and 2Molecular et al. 1999) and protein microarrays (MacBeath and and Cellular Biology Program, Campus Box 8069 Schreiber 2000; Zhu et al. 2000; Haab et al. 2001). In this Washington University School of Medicine article, we will provide a detailed analysis of the current 660 South Euclid Avenue, St. Louis, Missouri 63110 USA state of chemical genomics and its potential impact on biological and medical research, and pharmaceutical development. Abstract Chemical Biology or Genetics The knowledge of complete sequences of different organisms is dramatically changing the landscape of Since the seminal study of pea genetics by Mendal in 1865, biological research and pharmaceutical development. genetic analysis has been the benchmark for understanding We are experiencing a transition from a trial-and-error gene or protein functions. In classical genetics or forward approach in traditional biological research and natural genetics, the genomic DNA of a model organism or cell is product drug discovery to a systematic operation in randomly mutagenized to generate large numbers of genomics and target-specific drug design and mutants, which are screened for a desirable phenotype or selection. Small, cell-permeable and target-specific trait, such as alteration in growth, appearance or behavior. chemical ligands are particularly useful in systematic The phenotypes are then used to identify the responsible genomic approaches to study biological questions. -

Human Prion-Like Proteins and Their Relevance in Disease
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Universitat Autònoma de Barcelona Departament de Bioquímica i Biologia Molecular Institut de Biotecnologia i Biomedicina HUMAN PRION-LIKE PROTEINS AND THEIR RELEVANCE IN DISEASE Doctoral thesis presented by Cristina Batlle Carreras for the degree of PhD in Biochemistry, Molecular Biology and Biomedicine from the Universitat Autònoma de Barcelona. The work described herein has been performed in the Department of Biochemistry and Molecular Biology and in the Institute of Biotechnology and Biomedicine, supervised by Prof. Salvador Ventura i Zamora. Cristina Batlle Carreras Prof. Salvador Ventura i Zamora Bellaterra, 2020 Protein Folding and Conformational Diseases Lab. This work was financed with the fellowship “Formación de Profesorado Universitario” by “Ministerio de Ciencia, Innovación y Universidades”. This work is licensed under a Creative Commons Attributions-NonCommercial-ShareAlike 4.0 (CC BY-NC- SA 4.0) International License. The extent of this license does not apply to the copyrighted publications and images reproduced with permission. (CC BY-NC-SA 4.0) Batlle, Cristina: Human prion-like proteins and their relevance in disease. Doctoral Thesis, Universitat Autònoma de Barcelona (2020) English summary ENGLISH SUMMARY Prion-like proteins have attracted significant attention in the last years. -

Sequence Overlap Between Autosomal and Sex-Linked Probes on the Illumina Humanmethylation27 Microarray
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Genomics 97 (2011) 214–222 Contents lists available at ScienceDirect Genomics journal homepage: www.elsevier.com/locate/ygeno Sequence overlap between autosomal and sex-linked probes on the Illumina HumanMethylation27 microarray Yi-an Chen a,b, Sanaa Choufani a, Jose Carlos Ferreira a,b, Daria Grafodatskaya a, Darci T. Butcher a, Rosanna Weksberg a,b,⁎ a Program in Genetics and Genome Biology, Hospital for Sick Children Research Institute, Toronto, Ontario, Canada b Institute of Medical Science, University of Toronto, Toronto, Ontario, Canada article info abstract Article history: The Illumina Infinium HumanMethylation27 BeadChip (Illumina 27k) microarray is a high-throughput Received 12 August 2010 platform capable of interrogating the human DNA methylome. In a search for autosomal sex-specific DNA Accepted 18 December 2010 methylation using this microarray, we discovered autosomal CpG loci showing significant methylation Available online 4 January 2011 differences between the sexes. However, we found that the majority of these probes cross-reacted with sequences from sex chromosomes. Moreover, we determined that 6–10% of the microarray probes are non- Keywords: specific and map to highly homologous genomic sequences. Using probes targeting different CpGs that are Illumina Infinium HumanMethylation 27 BeadChip exact duplicates of each other, we investigated the precision of these repeat measurements and concluded Non-specific cross-reactive probe that the overall precision of this microarray is excellent. In addition, we identified a small number of probes Sex-specific DNA methylation targeting CpGs that include single-nucleotide polymorphisms. -

W Yang Et Al. Proteomic Approaches to the Analysis of Multiprotein
832 DOI 10.1002/pmic.200700650 Proteomics 2008, 8, 832–851 REVIEW Proteomic approaches to the analysis of multiprotein signaling complexes Wei Yang1, 2, Hanno Steen3 and Michael R. Freeman1, 2 1 The Urological Diseases Research Center, Department of Urology, Children’s Hospital Boston, Boston, MA, USA 2 Departments of Surgery, Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, USA 3 Department of Pathology, Harvard Medical School and Children’s Hospital Boston, Boston, MA, USA Signal transduction is one of the most active fields in modern biomedical research. Increasing Received: July 9, 2007 evidence has shown that signaling proteins associate with each other in characteristic ways to Revised: October 23, 2007 form large signaling complexes. These diverse structures operate to boost signaling efficiency, Accepted: October 23, 2007 ensure specificity and increase sensitivity of the biochemical circuitry. Traditional methods of protein analysis are inadequate to fully characterize and understand these structures, which are intricate, contain many components and are highly dynamic. Instead, proteomics technologies are currently being applied to investigate the nature and composition of multimeric signaling complexes. This review presents commonly used and potential proteomic methods of analyzing diverse protein complexes along with a discussion and a brief evaluation of alternative ap- proaches. Challenges associated with proteomic analysis of signaling complexes are also dis- cussed. Keywords: Cross-linking / Mass spectrometry / Post-translational modification / Quantitative proteomics / Signaling complexes 1 Introduction 1980s, astonishingly rapid progress has been made in understanding the mechanisms of signal transduction. The modern field of cell signaling can be traced back to the Many old concepts have been abandoned or revised and mid-1950s, when it was discovered that reversible phospho- new ones have emerged. -

Protein Interaction Network of Alternatively Spliced Isoforms from Brain Links Genetic Risk Factors for Autism
ARTICLE Received 24 Aug 2013 | Accepted 14 Mar 2014 | Published 11 Apr 2014 DOI: 10.1038/ncomms4650 OPEN Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism Roser Corominas1,*, Xinping Yang2,3,*, Guan Ning Lin1,*, Shuli Kang1,*, Yun Shen2,3, Lila Ghamsari2,3,w, Martin Broly2,3, Maria Rodriguez2,3, Stanley Tam2,3, Shelly A. Trigg2,3,w, Changyu Fan2,3, Song Yi2,3, Murat Tasan4, Irma Lemmens5, Xingyan Kuang6, Nan Zhao6, Dheeraj Malhotra7, Jacob J. Michaelson7,w, Vladimir Vacic8, Michael A. Calderwood2,3, Frederick P. Roth2,3,4, Jan Tavernier5, Steve Horvath9, Kourosh Salehi-Ashtiani2,3,w, Dmitry Korkin6, Jonathan Sebat7, David E. Hill2,3, Tong Hao2,3, Marc Vidal2,3 & Lilia M. Iakoucheva1 Increased risk for autism spectrum disorders (ASD) is attributed to hundreds of genetic loci. The convergence of ASD variants have been investigated using various approaches, including protein interactions extracted from the published literature. However, these datasets are frequently incomplete, carry biases and are limited to interactions of a single splicing isoform, which may not be expressed in the disease-relevant tissue. Here we introduce a new interactome mapping approach by experimentally identifying interactions between brain-expressed alternatively spliced variants of ASD risk factors. The Autism Spliceform Interaction Network reveals that almost half of the detected interactions and about 30% of the newly identified interacting partners represent contribution from splicing variants, emphasizing the importance of isoform networks. Isoform interactions greatly contribute to establishing direct physical connections between proteins from the de novo autism CNVs. Our findings demonstrate the critical role of spliceform networks for translating genetic knowledge into a better understanding of human diseases. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Produktinformation
Produktinformation Diagnostik & molekulare Diagnostik Laborgeräte & Service Zellkultur & Verbrauchsmaterial Forschungsprodukte & Biochemikalien Weitere Information auf den folgenden Seiten! See the following pages for more information! Lieferung & Zahlungsart Lieferung: frei Haus Bestellung auf Rechnung SZABO-SCANDIC Lieferung: € 10,- HandelsgmbH & Co KG Erstbestellung Vorauskassa Quellenstraße 110, A-1100 Wien T. +43(0)1 489 3961-0 Zuschläge F. +43(0)1 489 3961-7 [email protected] • Mindermengenzuschlag www.szabo-scandic.com • Trockeneiszuschlag • Gefahrgutzuschlag linkedin.com/company/szaboscandic • Expressversand facebook.com/szaboscandic MYCBP monoclonal antibody (M13), clone 1B12 Catalog # : H00026292-M13 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Transfected lysate) Description: MYCBP. Immunogen: MYCBP (NP_036465.2, 34 a.a. ~ 103 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: LYEEPEKPNSALDFLKHHLGAATPENPEIELLRLELAEMKEKYEAIVEENK KLKAKLAQYEPPQEEKRAE enlarge Host: Mouse Western Blot (Recombinant protein) Reactivity: Human Sandwich ELISA (Recombinant Isotype: IgG2a Kappa protein) Quality Control Antibody Reactive Against Recombinant Protein. Testing: enlarge ELISA Western Blot detection against Immunogen (33.44 KDa) . Storage Buffer: In 1x PBS, pH 7.4 Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. Instruction: MSDS: Download Datasheet: Download Applications Western Blot (Transfected lysate) Page 1 of 3 2016/5/23 Western Blot analysis of MYCBP expression in transfected 293T cell line by MYCBP monoclonal antibody (M13), clone 1B12. Lane 1: MYCBP transfected lysate(12 KDa). Lane 2: Non-transfected lysate. Protocol Download Western Blot (Recombinant protein) Protocol Download Sandwich ELISA (Recombinant protein) Detection limit for recombinant GST tagged MYCBP is 0.1 ng/ml as a capture antibody. -
Supplementary Table 2 Supplementary Table 1
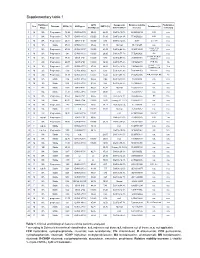
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Automethylation of PRC2 Promotes H3K27 Methylation and Is Impaired in H3K27M Pediatric Glioma
Downloaded from genesdev.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma Chul-Hwan Lee,1,2,7 Jia-Ray Yu,1,2,7 Jeffrey Granat,1,2,7 Ricardo Saldaña-Meyer,1,2 Joshua Andrade,3 Gary LeRoy,1,2 Ying Jin,4 Peder Lund,5 James M. Stafford,1,2,6 Benjamin A. Garcia,5 Beatrix Ueberheide,3 and Danny Reinberg1,2 1Department of Biochemistry and Molecular Pharmacology, New York University School of Medicine, New York, New York 10016, USA; 2Howard Hughes Medical Institute, Chevy Chase, Maryland 20815, USA; 3Proteomics Laboratory, New York University School of Medicine, New York, New York 10016, USA; 4Shared Bioinformatics Core, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724, USA; 5Department of Biochemistry and Molecular Biophysics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA The histone methyltransferase activity of PRC2 is central to the formation of H3K27me3-decorated facultative heterochromatin and gene silencing. In addition, PRC2 has been shown to automethylate its core subunits, EZH1/ EZH2 and SUZ12. Here, we identify the lysine residues at which EZH1/EZH2 are automethylated with EZH2-K510 and EZH2-K514 being the major such sites in vivo. Automethylated EZH2/PRC2 exhibits a higher level of histone methyltransferase activity and is required for attaining proper cellular levels of H3K27me3. While occurring inde- pendently of PRC2 recruitment to chromatin, automethylation promotes PRC2 accessibility to the histone H3 tail. Intriguingly, EZH2 automethylation is significantly reduced in diffuse intrinsic pontine glioma (DIPG) cells that carry a lysine-to-methionine substitution in histone H3 (H3K27M), but not in cells that carry either EZH2 or EED mutants that abrogate PRC2 allosteric activation, indicating that H3K27M impairs the intrinsic activity of PRC2. -

Essential Genes and Their Role in Autism Spectrum Disorder
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2017 Essential Genes And Their Role In Autism Spectrum Disorder Xiao Ji University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, and the Genetics Commons Recommended Citation Ji, Xiao, "Essential Genes And Their Role In Autism Spectrum Disorder" (2017). Publicly Accessible Penn Dissertations. 2369. https://repository.upenn.edu/edissertations/2369 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/2369 For more information, please contact [email protected]. Essential Genes And Their Role In Autism Spectrum Disorder Abstract Essential genes (EGs) play central roles in fundamental cellular processes and are required for the survival of an organism. EGs are enriched for human disease genes and are under strong purifying selection. This intolerance to deleterious mutations, commonly observed haploinsufficiency and the importance of EGs in pre- and postnatal development suggests a possible cumulative effect of deleterious variants in EGs on complex neurodevelopmental disorders. Autism spectrum disorder (ASD) is a heterogeneous, highly heritable neurodevelopmental syndrome characterized by impaired social interaction, communication and repetitive behavior. More and more genetic evidence points to a polygenic model of ASD and it is estimated that hundreds of genes contribute to ASD. The central question addressed in this dissertation is whether genes with a strong effect on survival and fitness (i.e. EGs) play a specific oler in ASD risk. I compiled a comprehensive catalog of 3,915 mammalian EGs by combining human orthologs of lethal genes in knockout mice and genes responsible for cell-based essentiality.