Supplementary Materials For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
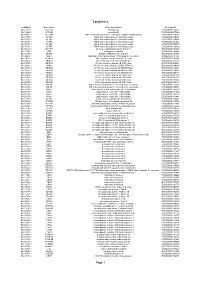
Epigenetics Page 1
Epigenetics esiRNA ID Gene Name Gene Description Ensembl ID HU-13237-1 ACTL6A actin-like 6A ENSG00000136518 HU-13925-1 ACTL6B actin-like 6B ENSG00000077080 HU-14457-1 ACTR1A ARP1 actin-related protein 1 homolog A, centractin alpha (yeast) ENSG00000138107 HU-10579-1 ACTR2 ARP2 actin-related protein 2 homolog (yeast) ENSG00000138071 HU-10837-1 ACTR3 ARP3 actin-related protein 3 homolog (yeast) ENSG00000115091 HU-09776-1 ACTR5 ARP5 actin-related protein 5 homolog (yeast) ENSG00000101442 HU-00773-1 ACTR6 ARP6 actin-related protein 6 homolog (yeast) ENSG00000075089 HU-07176-1 ACTR8 ARP8 actin-related protein 8 homolog (yeast) ENSG00000113812 HU-09411-1 AHCTF1 AT hook containing transcription factor 1 ENSG00000153207 HU-15150-1 AIRE autoimmune regulator ENSG00000160224 HU-12332-1 AKAP1 A kinase (PRKA) anchor protein 1 ENSG00000121057 HU-04065-1 ALG13 asparagine-linked glycosylation 13 homolog (S. cerevisiae) ENSG00000101901 HU-13552-1 ALKBH1 alkB, alkylation repair homolog 1 (E. coli) ENSG00000100601 HU-06662-1 ARID1A AT rich interactive domain 1A (SWI-like) ENSG00000117713 HU-12790-1 ARID1B AT rich interactive domain 1B (SWI1-like) ENSG00000049618 HU-09415-1 ARID2 AT rich interactive domain 2 (ARID, RFX-like) ENSG00000189079 HU-03890-1 ARID3A AT rich interactive domain 3A (BRIGHT-like) ENSG00000116017 HU-14677-1 ARID3B AT rich interactive domain 3B (BRIGHT-like) ENSG00000179361 HU-14203-1 ARID3C AT rich interactive domain 3C (BRIGHT-like) ENSG00000205143 HU-09104-1 ARID4A AT rich interactive domain 4A (RBP1-like) ENSG00000032219 HU-12512-1 ARID4B AT rich interactive domain 4B (RBP1-like) ENSG00000054267 HU-12520-1 ARID5A AT rich interactive domain 5A (MRF1-like) ENSG00000196843 HU-06595-1 ARID5B AT rich interactive domain 5B (MRF1-like) ENSG00000150347 HU-00556-1 ASF1A ASF1 anti-silencing function 1 homolog A (S. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Materials and Tables a and B
SUPPLEMENTARY MATERIAL 1 Table A. Main characteristics of the subset of 23 AML patients studied by high-density arrays (subset A) WBC BM blasts MYST3- MLL Age/Gender WHO / FAB subtype Karyotype FLT3-ITD NPM status (x109/L) (%) CREBBP status 1 51 / F M4 NA 21 78 + - G A 2 28 / M M4 t(8;16)(p11;p13) 8 92 + - G G 3 53 / F M4 t(8;16)(p11;p13) 27 96 + NA G NA 4 24 / M PML-RARα / M3 t(15;17) 5 90 - - G G 5 52 / M PML-RARα / M3 t(15;17) 1.5 75 - - G G 6 31 / F PML-RARα / M3 t(15;17) 3.2 89 - - G G 7 23 / M RUNX1-RUNX1T1 / M2 t(8;21) 38 34 - + ND G 8 52 / M RUNX1-RUNX1T1 / M2 t(8;21) 8 68 - - ND G 9 40 / M RUNX1-RUNX1T1 / M2 t(8;21) 5.1 54 - - ND G 10 63 / M CBFβ-MYH11 / M4 inv(16) 297 80 - - ND G 11 63 / M CBFβ-MYH11 / M4 inv(16) 7 74 - - ND G 12 59 / M CBFβ-MYH11 / M0 t(16;16) 108 94 - - ND G 13 41 / F MLLT3-MLL / M5 t(9;11) 51 90 - + G R 14 38 / F M5 46, XX 36 79 - + G G 15 76 / M M4 46 XY, der(10) 21 90 - - G NA 16 59 / M M4 NA 29 59 - - M G 17 26 / M M5 46, XY 295 92 - + G G 18 62 / F M5 NA 67 88 - + M A 19 47 / F M5 del(11q23) 17 78 - + M G 20 50 / F M5 46, XX 61 59 - + M G 21 28 / F M5 46, XX 132 90 - + G G 22 30 / F AML-MD / M5 46, XX 6 79 - + M G 23 64 / M AML-MD / M1 46, XY 17 83 - + M G WBC: white blood cell. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Supplementary Tables S1-S3
Supplementary Table S1: Real time RT-PCR primers COX-2 Forward 5’- CCACTTCAAGGGAGTCTGGA -3’ Reverse 5’- AAGGGCCCTGGTGTAGTAGG -3’ Wnt5a Forward 5’- TGAATAACCCTGTTCAGATGTCA -3’ Reverse 5’- TGTACTGCATGTGGTCCTGA -3’ Spp1 Forward 5'- GACCCATCTCAGAAGCAGAA -3' Reverse 5'- TTCGTCAGATTCATCCGAGT -3' CUGBP2 Forward 5’- ATGCAACAGCTCAACACTGC -3’ Reverse 5’- CAGCGTTGCCAGATTCTGTA -3’ Supplementary Table S2: Genes synergistically regulated by oncogenic Ras and TGF-β AU-rich probe_id Gene Name Gene Symbol element Fold change RasV12 + TGF-β RasV12 TGF-β 1368519_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 42.22 5.53 75.28 1373000_at sushi-repeat-containing protein, X-linked 2 (predicted) Srpx2 19.24 25.59 73.63 1383486_at Transcribed locus --- ARE 5.93 27.94 52.85 1367581_a_at secreted phosphoprotein 1 Spp1 2.46 19.28 49.76 1368359_a_at VGF nerve growth factor inducible Vgf 3.11 4.61 48.10 1392618_at Transcribed locus --- ARE 3.48 24.30 45.76 1398302_at prolactin-like protein F Prlpf ARE 1.39 3.29 45.23 1392264_s_at serine (or cysteine) peptidase inhibitor, clade E, member 1 Serpine1 ARE 24.92 3.67 40.09 1391022_at laminin, beta 3 Lamb3 2.13 3.31 38.15 1384605_at Transcribed locus --- 2.94 14.57 37.91 1367973_at chemokine (C-C motif) ligand 2 Ccl2 ARE 5.47 17.28 37.90 1369249_at progressive ankylosis homolog (mouse) Ank ARE 3.12 8.33 33.58 1398479_at ryanodine receptor 3 Ryr3 ARE 1.42 9.28 29.65 1371194_at tumor necrosis factor alpha induced protein 6 Tnfaip6 ARE 2.95 7.90 29.24 1386344_at Progressive ankylosis homolog (mouse) -

Supplemental Figure 1 Characterization of Lung Tumors In
Supplemental Figure 1 Characterization of lung tumors in KC mice. (A) Scatter plots of visible spontaneous tumors counted on the lung surfaces at necropsy. Each dot represents a value from a single mouse, and the bars and whiskers show the mean SD for each cohort from a single experiment. P value, two-tailed Student’s t-test. (B) Histologic analysis of spontaneous lung tumors in (A). Results indicate the numbers of lung lesions per histologic subtype per genotype. N, cohort sample sizes. P values, Fisher's exact test. Supplemental Figure 2 Surfactant protein C (SPC) expression in p21-deficient (KC) and –replete (K) tumor cell lines. Immunofluorescence staining with anti-surfactant protein C antibody (green) or normal IgG as a negative control. DAPI, blue. SPC/DAPI and IgG/DAPI are merged images. Scale bar, 20 µm. Supplemental Figure 3 Somatic recombination of latent KrasG12D allele and detection of p21 protein in p21-deficient (KC) and –replete (K) tumor cell lines. (A) PCR analysis of the K-rasLA1 and K-rasWT alleles in genomic DNA. Somatic recombination of the K-rasLA1 allele leads to loss of the PCR product (upper band, arrow). Wild-type K-ras allele detected (250 bp). Tail DNAs from KrasLA1 mice (KrasLA1/+) and wild-type mice (Kras+/+) were used as positive and negative controls, respectively, for the presence of the germline K-rasLA1 allele. (B) Immunoblot analysis of p21 expression. Actin was used as a loading control. Supplemental Figure 4 Growth and metastasis of p21-deficient (KC) tumor cell lines in Cdkn1a-null or –wild-type mice. -
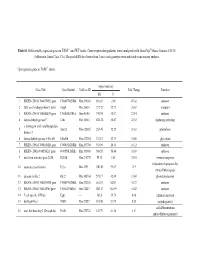
Table S1. Differentially Expressed Genes in TAM-/- and WT Testis. Gene Expression Patterns Were Analyzed with Genechip® Mouse
Table S1. Differentially expressed genes in TAM-/- and WT testis. Gene expression patterns were analyzed with GeneChip® Mouse Genome 430/2.0 (Affymetrix, Santa Clara, CA). The pooled RNAs of testes from 3 mice each genotype were subjected to microarray analysis. Up-regulated genes in TAM-/- testis. Signal intensity Gene Title Gene Symbol UniGene ID Fold Change Function KO N 1 RIKEN cDNA 1700097M23 gene 1700097M23Rik Mm.399360 116.83 2.48 47.02 unkown 2 fatty acid binding protein 9, testis Fabp9 Mm.26654 1219.27 45.72 26.67 transport 3 RIKEN cDNA 1700008K24 gene 1700008K24Rik Mm.46140 741.94 32.07 23.14 unkown 4 lactate dehydrogenase C Ldhc Mm.16563 1226.78 54.47 22.52 oxidation reduction a disintegrin and metallopeptidase 5 Adam2 Mm.422850 253.45 12.29 20.62 proteolysis domain 2 6 lactate dehydrogenase A-like 6B Ldhal6b Mm.432764 374.12 22.19 16.86 glycolysis 7 RIKEN cDNA 1700016M24 gene 1700016M24Rik Mm.263708 394.98 24.51 16.12 unkown 8 RIKEN cDNA 4930550L11 gene 4930550L11Rik Mm.391968 908.55 56.46 16.09 unkown 9 interferon activated gene 202B Ifi202b Mm.218770 55.32 3.63 15.24 immune response induction of apoptosis by 10 tumor necrosis factor α Tnf α Mm.1293 1383.03 99.47 13.9 extracellular signals 11 phosducin-like 2 Pdcl2 Mm.143764 571.17 42.49 13.44 phototransduction 12 RIKEN cDNA 1700029M20 gene 1700029M20Rik Mm.292326 665.07 62.07 10.72 unkown 13 RIKEN cDNA 1700102P08 gene 1700102P08Rik Mm.78287 1263.87 118.99 10.62 unkown 14 T-cell specific GTPase Tgtp --- 505.8 59.76 8.46 immune response 15 dickkopf-like 1 Dkkl1 Mm.27287 181.48 -

Supporting Information for Proteomics DOI 10.1002/Pmic.200500648
Supporting Information for Proteomics DOI 10.1002/pmic.200500648 Fiona M. McCarthy, Amanda M. Cooksey, Nan Wang, Susan M. Bridges, G. Todd Pharr and Shane C. Burgess Modeling a whole organ using proteomics: The avian bursa of Fabricius ª 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com Supporting Text: Determining Tissue Expression Patterns for Transcription Factors. We identified 107 transcription factors (TFs) from post-hatch bursas. Since the Transfac database (1) did not contain all of these TFs, we used PubMed literature searches to determine expression patterns for each. For each transcription factor, we searched PubMed for a combination of the protein and gene names, as determined by both NCBI and UniProt records. For example, Pax7 (gi:2576239; UniProt O42349) was searched as both PAX7 and Pax-7. In each case, articles were manually searched to confirm TF expression. We combined the TF names with the following terms to obtain reported tissue expression patterns: 1. Expression in the bursa. AND bursa* OR DT40 eg. (Pax7 OR “Pax-7”) AND (bursa* OR DT40) returned no items. 2. Expression in immune tissue. AND lymphocyte* OR leukocyte* OR “bone marrow” OR spleen OR “embryonic liver” OR hematopoietic eg. (Pax7 OR “Pax-7”) AND (lymphocyte* OR leukocyte* OR “bone marrow” OR spleen OR “embryonic liver” OR hematopoietic) returned 17 articles. 3. Expression during development. AND development OR embryo* eg. (Pax7 OR “Pax-7”) AND (development OR embryo*) returned 135 articles. 4. Expression in nervous tissue. AND neuron* OR nervous OR CNS OR retina* 1 eg. (Pax7 OR “Pax-7”) AND (neuron* OR nervous OR CNS OR retina*) returned 53 articles. -

Eudragit® RL) Nanoparticles by Human THP-1 Cell Line and Its Effects on Hematology and Erythrocyte Damage in Rats
AVERTISSEMENT Ce document est le fruit d'un long travail approuvé par le jury de soutenance et mis à disposition de l'ensemble de la communauté universitaire élargie. Il est soumis à la propriété intellectuelle de l'auteur. Ceci implique une obligation de citation et de référencement lors de l’utilisation de ce document. D'autre part, toute contrefaçon, plagiat, reproduction illicite encourt une poursuite pénale. Contact : [email protected] LIENS Code de la Propriété Intellectuelle. articles L 122. 4 Code de la Propriété Intellectuelle. articles L 335.2- L 335.10 http://www.cfcopies.com/V2/leg/leg_droi.php http://www.culture.gouv.fr/culture/infos-pratiques/droits/protection.htm Ecole Doctorale BioSE (Biologie-Santé-Environnement) Thèse Présentée et soutenue publiquement pour l’obtention du titre de DOCTEUR DE l’UNIVERSITE DE LORRAINE Mention : « Sciences de la Vie et de la Santé » par Ramia SAFAR Etude toxicologique de nanoparticules polymériques véhicules de S-nitrosoglutathion Le 4 décembre 2015 Membres du jury : Rapporteurs : Mme Armelle BAEZA Professeur, Université Paris Diderot, Paris Mme Françoise PONS Professeur, Université de Strasbourg, Strasbourg Examinateurs : M. Bertrand RIHN Professeur, Université de Lorraine, Nancy Directeur de thèse M. Olivier JOUBERT Maître de conférences, Université de Lorraine, Nancy Co-directeur de thèse Invités : M. Jean-Claude ANDRE Directeur de Recherche Emérite, INSIS CNRS, Paris M. Alain LE FAOU Professeur Emérite, Université de Lorraine, Nancy EA 3452 CITHEFOR, « Cibles Thérapeutiques, -

Hormone Induced Differential Transcriptome Analysis of Sertoli Cells During Postnatal Maturation of Rat Testes
RESEARCH ARTICLE Hormone induced differential transcriptome analysis of Sertoli cells during postnatal maturation of rat testes Mukesh Gautam1☯¤a, Indrashis Bhattacharya2☯¤b, Umesh Rai1, Subeer S. Majumdar2,3* 1 Department of Zoology, University of Delhi, Delhi, India, 2 Cellular Endocrinology Laboratory, National Institute of Immunology, New Delhi, India, 3 National Institute of Animal Biotechnology, Hyderabad, India ☯ These authors contributed equally to this work. a1111111111 ¤a Current address: The Ken & Ruth Davee Department of Neurology, Northwestern University, Chicago, a1111111111 Illinois, United States of America a1111111111 ¤b Current address: Department of Zoology, Hemvati Nandan Bahuguna Garhwal University, Srinagar, a1111111111 Garhwal, Uttarakhand, India a1111111111 * [email protected] Abstract OPEN ACCESS Sertoli cells (Sc) are unique somatic cells of testis that are the target of both FSH and testos- Citation: Gautam M, Bhattacharya I, Rai U, terone (T) and regulate spermatogenesis. Although Sc of neonatal rat testes are exposed to Majumdar SS (2018) Hormone induced differential high levels of FSH and T, robust differentiation of spermatogonial cells becomes conspicu- transcriptome analysis of Sertoli cells during ous only after 11-days of postnatal age. We have demonstrated earlier that a developmental postnatal maturation of rat testes. PLoS ONE 13(1): e0191201. https://doi.org/10.1371/journal. switch in terms of hormonal responsiveness occurs in rat Sc at around 12 days of postnatal pone.0191201 age during the rapid transition of spermatogonia A to B. Therefore, such ªfunctional matura- Editor: Suresh Yenugu, University of Hyderabad, tionº of Sc, during pubertal development becomes prerequisite for the onset of spermato- INDIA genesis. However, a conspicuous difference in robust hormone (both T and FSH) induced Received: August 20, 2017 gene expression during the different phases of Sc maturation restricts our understanding about molecular events necessary for the spermatogenic onset and maintenance. -

Detection of Tissue-Specific Genes and Computational Analysis of Testis
Detection of tissue-specific genes and Title computational analysis of testis-specific gene expression regulatory regions Author(s) 山下, 明史 Citation Issue Date Text Version ETD URL http://hdl.handle.net/11094/2599 DOI rights Note Osaka University Knowledge Archive : OUKA https://ir.library.osaka-u.ac.jp/ Osaka University Detection of tissue-specific genes and computational analysis of testis-specific gene expression regulatory regions (組織特異的遺伝子の検出と精巣特異的遺伝子の 発現制御領域のコンピュータを使った解析) by Akifumi Yamashita Department of Genome Informatics, Genome Information Research Center, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan February, 2009 1/97 Contents LIST of ABBREVIATIONS 5 ABSTRACT 6 1 INTRODUCTION 7 2 METHODS 10 2.1 Detection of tissue-specific genes 10 2.2 Annotation of tissue-specific genes 10 2.3 Extracting the 5′-regulatory regions 11 2.4 Selection of over-represented 8-mers 11 2.5 Comparison of the 8-mer frequency within regulatory regions of 13 testis-specific genes with those of non-testis-specific genes. 3 RESULTS 14 3.1 Detection of tissue-specific genes 14 3.2 Selection of testis-specific genes which can be used for the 14 following analysis 3.3 Classification of the over-represented 8-mers 15 3.4 Conservations of the flanking region of highlighted 8-mers 16 2/97 4 DISCUSSION 18 ACKNOWLEDGEMENTS 23 REFERENCES 24 TABLES 27 Table 1 - Number of tissue-specific genes 27 Table 2 - Classification of 117 representative 8-mers on the basis of their 28 testis association level Table 3 - 8-mers which appeared near by the other 8-mers 31 FIGURES 32 Fig.