Azido-2',3'-Dideoxynucleoside Analog Reverse Transcriptase Inhibi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
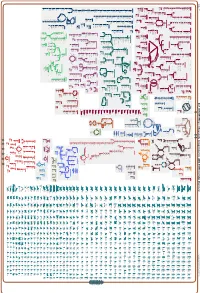
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

The Clinical Biochemistry of 5'-Nucleotidase*
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 20, No. 2 Copyright © 1990, Institute for Clinical Science, Inc. The Clinical Biochemistry of 5'-Nucleotidase* F. WILLIAM SUNDERMAN JR., M.D. Departments of Laboratory Medicine and Pharmacology, University of Connecticut Medical School, Farmington, CT 06032 ABSTRACT This review delineates the subcellular distribution, biochemical charac teristics, and metabolic functions of 5'-nucleotidase (5'NT), summarizes the analytical biochemistry of 5'NT, and assesses the clinical significance of5'NT determinations in body fluids, cells, and tissues. Salient aspects of the clinical biochemistry of 5'NT, discussed herein, are as follows: (A) Serum 5'NT activity is generally elevated in hepatobiliary diseases, espe cially with intrahepatic obstruction, but, unlike serum alkaline phospha tase, serum 5'NT activity is not increased in infancy, childhood, preg nancy, or osteoblastic disorders. (B) In cancer patients, elevated serum 5'NT activity does not always indicate hepatobiliary involvement; in some cases, 5'NT may be released into serum from the primary tumor or local metastases. (C) Genetic deficiency of erythrocyte pyrimidine 5'NT activity is a common cause of hereditary non-spherocytic hemolytic anemia. (D) Acquired deficiency of erythrocyte pyrimidine 5'NT activity occurs in patients with P-thalassemia and lead poisoning. (E) 5'NT activity is low in circulating monocytes, increases markedly upon their differentiation to tissue macrophages, and subsequently diminishes during macrophage activation. (F) Lymphocyte ecto-5'NT activity, a plasma membrane marker of cell maturation, is generally low in immunodeficiency states, and undergoes characteristic changes in patients with certain lymphomas and leukemias. Introduction ribonucleosides and inorganic phos phate. -

Poly (I)-Poly (C)/Oligo (Dt)-Cellulosei... Sidney Pestka, James Mcinnes
Contents Vol. 72, No. 10 October 1975 INFORMATION TO CONTRIBUTORS ..............................iv...........................I..................... i-i AUTfHOR INDEX ................................................................................................. 4190 Physical Sciences APPLIED Adiabatic evolution of plasma equilibrium (bifurcation/isolation/weak solution/generalized differential MATHEMATICS equation/Tokamak) ............................................ H. Grad, P.N. Hu, and D. C. Stevens 3789-3793 CHEMISTRY Theoretical studies of metal-phosphate interactions: Interaction of Li+, Na+, K+, Be++, Mg++, and Ca++ with H2PO4- and (CH30)2PO2-: Implications for nucleic acid solvation (metal binding/phos- phate complexes/molecular orbital theory) ................Dennis S. Marynick and Henry F. Schaefer III 3794-3798 Maximum-valence radii of transition metals (covalent radii/enneacovalence) ............... Linus Pauling 3799-3801 Model of protein folding: Inclusion of short-, medium-, and long-range interactions (mechanism offolding/ contact free energies/range of interactions/Monte Carlo) ........... Seiji Tanaka and HaroldA. Scheraga 3802-3806 Matrix method for fhActuations and noise in kinetic systems (cross correlation function/noise power spectrum matrix/relaxation matrix/biochemical reaction kinetics/muscle contraction) .Yi-der Chen 3807-3811 MATHEMATICS Point estimates for probability moments (approximate methods/finite differences/numerical methods/prob- abilities) .......................... - ;Emilio Rosenblueth 3812-3814 -

Phosphodiesterases Expression During Murine Cardiac Development
International Journal of Molecular Sciences Article Phosphodiesterases Expression during Murine Cardiac Development Thays Maria da Conceição Silva Carvalho 1,†, Silvia Cardarelli 1,†, Mauro Giorgi 2, Andrea Lenzi 3, Andrea M. Isidori 3 and Fabio Naro 1,* 1 Department of Anatomical, Histological, Forensic and Orthopedic Sciences, Sapienza University, 00161 Rome, Italy; [email protected] (T.M.d.C.S.C.); [email protected] (S.C.) 2 Department of Biology and Biotechnology “C. Darwin”, Sapienza University, 00185 Rome, Italy; [email protected] 3 Department of Experimental Medicine, Sapienza University, 00161 Rome, Italy; [email protected] (A.L.); [email protected] (A.M.I.) * Correspondence: [email protected] † These authors contributed equally to this work. Abstract: 30-50 cyclic nucleotide phosphodiesterases (PDEs) are a large family of enzymes playing a fundamental role in the control of intracellular levels of cAMP and cGMP. Emerging evidence suggested an important role of phosphodiesterases in heart formation, but little is known about the expression of phosphodiesterases during cardiac development. In the present study, the pattern of expression and enzymatic activity of phosphodiesterases was investigated at different stages of heart formation. C57BL/6 mice were mated and embryos were collected from 14.5 to 18.5 days of development. Data obtained by qRT-PCR and Western blot analysis showed that seven different isoforms are expressed during heart development, and PDE1C, PDE2A, PDE4D, PDE5A and PDE8A Citation: Carvalho, T.M.d.C.S.; are modulated from E14.5 to E18.5. In heart homogenates, the total cAMP and cGMP hydrolytic Cardarelli, S.; Giorgi, M.; Lenzi, A.; activity is constant at the evaluated times, and PDE4 accounts for the majority of the cAMP hydrolyz- Isidori, A.M.; Naro, F. -

Oxidative Stress Responses in Escherichia Coli and Salmonella Typhimurium
MICROBIOLOGICAL REVIEWS, Dec. 1991, p. 561-585 Vol. 55, No. 4 0146-07 49/91/040561-25$02. 00/0 Copyright © 1991, American Society for Microbiology Oxidative Stress Responses in Escherichia coli and Salmonella typhimurium SPENCER B. FARR1* AND TOKIO KOGOMA2 Department of Molecular and Cellular Toxicology, Harvard School of Public Health, Boston, Massachusetts 02115, 1 and Departments ofCell Biology and Microbiology, University of New Mexico Medical Center, Albuquerque, New Mexico 87131 2 INTRODUCTION AND SCOPE OF REVIEW ............................................................................ 561 OXIDATIVE STRESS RESPONSES .........................................................................................562 Active Oxygen Species ......................................................................................................... 562 Reactivities .....••••.•.......................•............................•.........•.......................................... 562 Sources ......................................•................•.......•.•.•..................................••...••....•.......563 Oxidative Stress and Cellular Responses .........................................................•.......•..•••••....•...563 Peroxide stress response •................•.......•.............................................................•...........564 Su peroxide stress response ...............•..........•.•........•....••....••.....................••.•••••...••.•..•..•..•.564 The two oxidative stress responses are distinct .......................................................................564 -

Bkca-Mediated PDE Regulation of Artery Tone
BKCa-mediated PDE regulation of artery tone Contribution of BKCa channels to vascular tone regulation by PDE3 and PDE4 is lost in heart failure Sarah Idres, Germain Perrin, Valérie Domergue, Florence Lefebvre, Susana Gomez, Audrey Varin, Rodolphe Fischmeister, Véronique Leblais, Boris Manoury To cite this version: Sarah Idres, Germain Perrin, Valérie Domergue, Florence Lefebvre, Susana Gomez, et al.. BKCa- mediated PDE regulation of artery tone Contribution of BKCa channels to vascular tone regulation by PDE3 and PDE4 is lost in heart failure. Cardiovascular Research, Oxford University Press (OUP), 2019, 115 (1), pp.130-144. 10.1093/cvr/cvy161. hal-02463726 HAL Id: hal-02463726 https://hal.archives-ouvertes.fr/hal-02463726 Submitted on 1 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Manuscript Idres et al., BKCa-mediated PDE regulation of artery tone MS # CVR-2017-669 Contribution of BKCa channels to vascular tone regulation by PDE3 and PDE4 is lost in heart failure Sarah Idres1, Germain Perrin1, Valérie Domergue2, Florence Lefebvre1, Susana Gomez1, Audrey Varin1, Rodolphe Fischmeister1, Véronique Leblais1 and Boris Manoury1. 1 Signalling and cardiovascular pathophysiology - UMR-S 1180, Univ. Paris-Sud, INSERM, Université Paris-Saclay, 92296, Châtenay-Malabry, France 2 UMS IPSIT, Univ. -

Nucleotidase) Up-Regulation ′ CD73
IFN-α Induced Adenosine Production on the Endothelium: A Mechanism Mediated by CD73 (Ecto-5′-Nucleotidase) Up-Regulation This information is current as Jussi Niemelä, Tiina Henttinen, Gennady G. Yegutkin, Laura of September 29, 2021. Airas, Anna-Maija Kujari, Pertti Rajala and Sirpa Jalkanen J Immunol 2004; 172:1646-1653; ; doi: 10.4049/jimmunol.172.3.1646 http://www.jimmunol.org/content/172/3/1646 Downloaded from References This article cites 41 articles, 21 of which you can access for free at: http://www.jimmunol.org/content/172/3/1646.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 29, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2004 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology IFN-␣ Induced Adenosine Production on the Endothelium: (A Mechanism Mediated by CD73 (Ecto-5-Nucleotidase Up-Regulation1 Jussi Niemela¨,* Tiina Henttinen,* Gennady G. Yegutkin,* Laura Airas,* Anna-Maija Kujari,* Pertti Rajala,† and Sirpa Jalkanen2* CD73 (ecto-5-nucleotidase; EC 3.1.3.5) participates in lymphocyte binding to endothelial cells and converts extracellular AMP into a potent anti-inflammatory substance adenosine. -

Disorder Index
Disorder Index A AIP. See Acute intermittent porphyria (AIP) AARF domain-containing kinase 3 (ADCK3) defi ciency , 235 AKU. See Alkaptonuria (AKU) ABCB4 defi ciency , 557, 559, 564, 571 Alanine-glyoxylate aminotransferase (AGT) , 376, 466, 467, 471, ABCB11 defi ciency , 557, 564, 571 763, 772 ABCC2 defi ciency , 557, 564 Alanine-glyoxylate aminotransferase defi ciency (peroxisomal) , Abetalipoproteinemia (MTP) , 674, 678 467, 763 ABS. See Antley-Bixler syndrome (ABS) ALDOA defi ciency. See Aldolase A (ALDOA) defi ciency ACAD9 defi ciency. See Acyl-CoA dehydrogenase 9 (ACAD9) Aldoketoreductase 2 defi ciency, Backdoor pathway defect , 605 defi ciency Aldolase A (ALDOA) defi ciency , 269, 821 N -Acetyl-alpha- d -glucosaminidase defi ciency , 450 Aldolase B (ALDOB) defi ciency , 268, 820 N -Acetylaspartic aciduria , 143 Aldosterone synthase defi ciency , 605 Acetyl-CoA alpha-glucosaminide acetyltransferase defi ciency , 450 ALG1-CDG. See Mannosyltransferase 1 defi ciency congenital N -Acetylgalactosamine-4-sulphatase defi ciency , 450 disorders of glycosylation (ALG1-CDG) N -Acetylgalactosamine-6-sulphatase defi ciency , 450 ALG 2-CDG. See Mannosyltransferase 2 defi ciency congenital N -Acetylglucosamine-1-phosphotransferase (alpha/beta) disorders of glycosylation (ALG 2-CDG) defi ciency , 402 ALG3-CDG. See Mannosyltransferase 6 defi ciency congenital N -Acetylglucosamine-1-phosphotransferase (gamma) defi ciency , 402 disorders of glycosylation (ALG3-CDG) N -Acetylglucosamine-6-sulphatase defi ciency , 450 ALG6-CDG. See Glucosyltransferase 1 defi ciency, congenital N -Acetylglucosaminyltransferase 2 congenital defects of glycosylation disorders of glycosylation (ALG6-CDG) (MGAT2-CDG) , 484–486, 489 ALG 8-CDG. See Glucosyltransferase 2 defi ciency, congenital N -Acetylglucosaminyltransferase 2 defi ciency-CDG-IIa , 486, 496 disorders of glycosylation (ALG 8-CDG) N -Acetylglutamate synthase defi ciency , 49, 51, 715, 818 ALG9-CDG. -

Introduction the Terms in the Subject Index for Volume 81, January
Introduction The terms in the Subject Index for Volume 81, January-December 1984, of the PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES USA (Biological Sci- ences) were chosen from titles, key terms, and abstracts of articles. The index terms are alphabetized by computer; numbers, conformational prefixes, and hy- phenated Greek letters are disregarded in alphabetization. 8041 8042 Biological Sciences Subject Index Proc. Natl. Acad. Sci. USA 81 (1984) A* protein Different channel properties of Torpedo acetylcholine receptor monomers Cleavage of single-stranded DNA by the <X174 A* protein: The A*- and dimers reconstituted in planar membranes, 6222 single-stranded DNA covalent linkage, 4285 Phosphorylation of the nicotinic acetylcholine receptor by an endogenous Abelson murine leukemia virus tyrosine-specific protein kinase, 6968 Transformation-associated proteins in murine B-cell lymphomas that are Purification of the muscarinic acetylcholine receptor from porcine atria distinct from Abelson virus gene products, 4434 (Correction), 7258 Abortive infection Acetylcholine receptor-specific suppressive T-cell factor from a Virus-plasmid interactions: Mutants of bacteriophage T3 that abortively retrovirally transformed T-cell line, 7569 infect plasmid F-containing (F+) strains of Escherichia coli, 1465 Isolation and characterization of a cDNA clone for the complete protein Cytomegalovirus infects human lymphocytes and monocytes: Virus coding region of the 8 subunit of the mouse acetylcholine expression is restricted to immediate-early gene products, -

Supplementary Information For
1 2 3 4 5 6 Supplementary information for: 7 5-Deoxyadenosine Salvage by Promiscuous Enzyme 8 Activity leads to Bioactive Deoxy-Sugar Synthesis in 9 Synechococcus elongatus 10 Johanna Rapp, Pascal Rath, Joachim Kilian, Klaus Brilisauer, Stephanie Grond, Karl 11 Forchhammer 1 12 Results 13 14 Figure S1: Only small intracellular 5dR and 7dSh accumulation in S. elongatus. Concentration -1 -1 15 of 5dR (black dots) and 7dSh (grey squares) in S. elongatus cells [µmol*L culture*OD750 ] 16 aerated with air supplemented with 2 % CO2. 2 17 18 Figure S2: 5dR-1P accumulates in crude extracts of S. elongatus that were incubated in the 19 presence of phosphate. Accumulation of 5dR-1P shown as extracted ion chromatogram 20 [M+H, M+Na]+ (m/z 215.0315; m/z 237.0135) in crude extracts of S. elongatus, (Red – with 21 5dAdo+potassium phosphate buffer (PPB); green – with 5dAdo, no PPB; black – without 22 5dAdo, PPB). Three independent replicates are shown for each treatment. One part of the 23 samples of the crude extract assays was analysed via high resolution LC-MS (C18 Gemini, 24 solvent A: ACN+0.1 %TFA, solvent B: H2O, 1% - 20% B in 20 min, Maxis 4G ESI-QTOF). 3 25 26 Figure S3: 5dR does not accumulate in crude extracts which were incubated with 5dAdo. 27 Crude extracts from S. elongatus or S. elongatus mtnP::specR were incubated with 5dAdo in 28 the presence or absence of potassium phosphate buffer (PPB) and then analysed via thin layer 29 chromatography (TLC). TLC plate from Figure 7 (main text) was sprayed with anisaldehyde 30 after UV-visualisation. -

Trna Biology Charges to the Front
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW tRNA biology charges to the front Eric M. Phizicky1,4 and Anita K. Hopper2,3 1Department of Biochemistry and Biophysics, Center for RNA Biology, University of Rochester School of Medicine, Rochester, New York 14642, USA; 2Department of Molecular Genetics, Center for RNA Biology, Ohio State University, Columbus, Ohio 43210, USA tRNA biology has come of age, revealing an unprecedented Multiple layers of regulation of tRNA transcription level of understanding and many unexpected discoveries tRNA and rRNA genes are highly transcribed, leading to along the way. This review highlights new findings on the the production in yeast of ;3 million tRNAs per gener- diverse pathways of tRNA maturation, and on the forma- ation and 300,000 ribosomes (Waldron and Lacroute tion and function of a number of modifications. Topics of 1975), compared with about 60,000 mRNAs (Ares et al. special focus include the regulation of tRNA biosynthesis, 1999). Because of the energy devoted to tRNA and rRNA quality control tRNA turnover mechanisms, widespread transcription, and because of the required coordination of tRNA cleavage pathways activated in response to stress tRNA and ribosome function, tRNA transcription via and other growth conditions, emerging evidence of signal- RNA polymerase III (Pol III) and rRNA transcription via ing pathways involving tRNA and cleavage fragments, and Pol I need to be coordinated and regulated in response to the sophisticated intracellular tRNA trafficking that oc- cellular nutrient availability and other environmental curs during and after biosynthesis. information. The consequences of inappropriate regula- tion of tRNA transcription have been underscored by the tRNA biogenesis involves the synthesis of the initial results of Marshall et al. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses by a Synthetic TLR3 Ligand Mitigates
Downloaded from http://www.jimmunol.org/ by guest on September 28, 2021 Inducing is online at: average * The Journal of Immunology published online 16 August 2013 from submission to initial decision 4 weeks from acceptance to publication http://www.jimmunol.org/content/early/2013/08/16/jimmun ol.1300376 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 28, 2021. Published August 16, 2013, doi:10.4049/jimmunol.1300376 The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN.