Identification and Characterization of Novel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Zebrafish Behavioral Profiling Links Drugs to Biological Targets and Rest/Wake Regulation
www.sciencemag.org/cgi/content/full/327/5963/348/DC1 Supporting Online Material for Zebrafish Behavioral Profiling Links Drugs to Biological Targets and Rest/Wake Regulation Jason Rihel,* David A. Prober, Anthony Arvanites, Kelvin Lam, Steven Zimmerman, Sumin Jang, Stephen J. Haggarty, David Kokel, Lee L. Rubin, Randall T. Peterson, Alexander F. Schier* *To whom correspondence should be addressed. E-mail: [email protected] (A.F.S.); [email protected] (J.R.) Published 15 January 2010, Science 327, 348 (2010) DOI: 10.1126/science.1183090 This PDF file includes: Materials and Methods SOM Text Figs. S1 to S18 Table S1 References Supporting Online Material Table of Contents Materials and Methods, pages 2-4 Supplemental Text 1-7, pages 5-10 Text 1. Psychotropic Drug Discovery, page 5 Text 2. Dose, pages 5-6 Text 3. Therapeutic Classes of Drugs Induce Correlated Behaviors, page 6 Text 4. Polypharmacology, pages 6-7 Text 5. Pharmacological Conservation, pages 7-9 Text 6. Non-overlapping Regulation of Rest/Wake States, page 9 Text 7. High Throughput Behavioral Screening in Practice, page 10 Supplemental Figure Legends, pages 11-14 Figure S1. Expanded hierarchical clustering analysis, pages 15-18 Figure S2. Hierarchical and k-means clustering yield similar cluster architectures, page 19 Figure S3. Expanded k-means clustergram, pages 20-23 Figure S4. Behavioral fingerprints are stable across a range of doses, page 24 Figure S5. Compounds that share biological targets have highly correlated behavioral fingerprints, page 25 Figure S6. Examples of compounds that share biological targets and/or structural similarity that give similar behavioral profiles, page 26 Figure S7. -

Annual Report 2013
Annual Report 2013 “ Being active and having a positive outlook on life is what keeps me going every day.” Overview of 2013 “ Our performance in 2013 was defined by remarkable &R D output and further delivery of sustained financial performance for our shareholders.” Please go to page 4 for more More at gsk.com Performance highlights £26.5bn £8.0bn £7.0bn £5.2bn Group turnover Core* operating profit Total operating profit Returned to shareholders 6 112.2p 112.5p 13% Major medicines approved Core* earnings per share Total earnings per share Estimated return on R&D investment 10 6 1st 1st Potential phase III study starts in 2014/15 Potential medicines with phase III data in Access to Medicines Index Pharmaceutical company to sign AllTrials expected 2014/15 campaign for research transparency Front cover story Betty, aged 65, (pictured) has Chronic “ Health is important to me, Obstructive Pulmonary Disease (COPD). She only has 25% lung capacity. This means I try to take care of my she finds even everyday tasks difficult, but medicines and inhaled oxygen allow her to health with all the tools live as normal a life as she can. Betty’s mindset I have and do the best is to stay busy and active, so every week she goes to rehab exercise classes. that I can with it.” COPD is a disease of the lungs that leads to Betty, COPD patient, damaged airways, causing them to become North Carolina, USA narrower and making it harder for air to get in and out. 210 million people around the world are estimated to have COPD. -

Other Statutory Disclosures Continued
Other statutory disclosures continued Strategic reportGroup companies Governance & remuneration Financial statements In accordance with Section 409 of the Companies Act 2006 a full list of subsidiaries, associates, joint ventures and joint arrangements, the country of incorporation and effective percentage of equity owned, as at 31 December 2015 are disclosed below. Unless otherwise stated the share capital disclosed comprises ordinary shares which are indirectly held by GlaxoSmithKline plc. All subsidiary companies are resident for tax purposes in their country of incorporation unless otherwise stated. Country of Effective % % Held by Name incorporation Ownership Security Class of Share Wholly owned subsidiaries 1506369 Alberta ULC Canada 100 Common 100 Action Potential Venture Capital Limited England & Wales 100 Ordinary 100 Adechsa GmbH Switzerland 100 Ordinary 100 Affymax Research Institute United States 100 Common 100 Alenfarma – Especialidades Farmaceuticas, Limitada (iv) Portugal 100 Ordinary Quota 100 Allen & Hanburys Limited (iv) England & Wales 100 Ordinary 100 Allen & Hanburys Pharmaceutical Nigeria Limited Nigeria 100 Ordinary 100 Allen Farmaceutica, S.A. Spain 100 Ordinary 100 Allen Pharmazeutika Gesellschaft m.b.H. Austria 100 Ordinary 100 Aners S.A (iv) Argentina 100 Non-endorsable Nominative Ordinary 100 Barrier Therapeutics, Inc. United States 100 Common 100 Beecham Group p l c England & Wales 100 20p Shares 'A'; 5p Shares B 100 Beecham Pharmaceuticals (Pte) Limited Singapore 100 Ordinary 100 Beecham Pharmaceuticals S.A (iv) (vi) Ecuador 100 Nominative 100 Beecham Portuguesa-Produtos Farmaceuticos e Quimicos, Lda Portugal 100 Ordinary Quota 100 Beecham S.A. (iv) Belgium 100 Ordinary 100 Biddle Sawyer Limited India 100 Equity 100 Biovesta Ilaçlari Ltd. Sti. Turkey 100 Nominative 100 Burroughs Wellcome & Co (Australia) Pty Limited (iv) (vi) Australia 100 Ordinary 100 Burroughs Wellcome & Co (Bangladesh) Limited Bangladesh 100 Ordinary 100 Burroughs Wellcome International Limited England & Wales 100 Ordinary 100 Caribbean Chemical Company, Ltd. -

Clinical Guidelines for Use of Depot Buprenorphine (Buvidal®
Clinical guidelines for use of depot buprenorphine (Buvidal® and Sublocade®) in the treatment of opioid dependence NSW Ministry of Health 100 Christie Street ST LEONARDS NSW 2065 Tel. (02) 9391 9000 Fax. (02) 9391 9101 TTY. (02) 9391 9900 www.health.nsw.gov.au Produced by: NSW Ministry of Health Lintzeris N, Dunlop A, Masters D (2019) Clinical guidelines for use of depot buprenorphine (Buvidal® and Sublocade®) in the treatment of opioid dependence. NSW Ministry of Health, Sydney Australia This work is copyright. It may be reproduced in whole or in part for study or training purposes subject to the inclusion of an acknowledgement of the source. It may not be reproduced for commercial usage or sale. Reproduction for purposes other than those indicated above requires written permission from the NSW Ministry of Health. © NSW Ministry of Health 2019 SHPN (CPH) 190431 ISBN 978-1-76081-226-3 Further copies of this document can be downloaded from the NSW Health website www.health.nsw.gov.au August 2019 ii NSW Health Use of depot BPN in the treatment of opioid dependence Authors Professor Nicholas Lintzeris (BMedSci MBBS PhD FAChAM) Director Drug and Alcohol Services, South East Sydney Local Health District Discipline Addiction Medicine, Faculty of Medicine and Health Sciences, University of Sydney NSW Drug and Alcohol Clinical Research and Improvement Network Professor Adrian Dunlop (MBBS, PhD, GdipEpiBiotat, FAChAM) Director Drug and Alcohol Clinical Services, Hunter New England Local Health District School of Medicine and Public Health, Faculty -

Glaxosmithkline Plc Annual Report for the Year Ended 31St December 2000
GlaxoSmithKline 01 GlaxoSmithKline plc Annual Report for the year ended 31st December 2000 Contents Report of the Directors 02 Financial summary 03 Joint statement by the Chairman and the Chief Executive Officer 05 Description of business 29 Corporate governance 37 Remuneration report 47 Operating and financial review and prospects 69 Financial statements 70 Directors’ statements of responsibility 71 Report by the auditors 72 Consolidated statement of profit and loss 72 Consolidated statement of total recognised gains and losses 74 Consolidated statement of cash flow 76 Consolidated balance sheet 76 Reconciliation of movements in equity shareholders’ funds 77 Company balance sheet 78 Notes to the financial statements 136 Group companies 142 Principal financial statements in US$ 144 Financial record 153 Investor information 154 Shareholder return 156 Taxation information for shareholders 157 Shareholder information 158 Share capital 160 Cross reference to Form 20-F 162 Glossary of terms The Annual Report was approved by the Board 163 Index of Directors on 22nd March 2001 and published on 12th April 2001. Contact details 02 GlaxoSmithKline Financial summary 2000 1999 Increase Business performance £m £m CER % £ % Sales 18,079 16,164 9 12 Trading profit 5,026 4,378 12 15 Profit before taxation 5,327 4,708 11 13 Earnings/Net income 3,697 3,222 13 15 Earnings per Ordinary Share 61.0p 52.7p 14 16 Total results Profit before taxation 6,029 4,236 Earnings/Net income 4,154 2,859 Earnings per Ordinary Share 68.5p 46.7p Business performance: results exclude merger items and restructuring costs; 1999 sales and trading profit exclude the Healthcare Services businesses which were disposed of in 1999. -
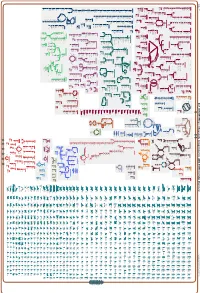
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_003855395Cyc: Shewanella livingstonensis LMG 19866 Cellular Overview Connections between pathways are omitted for legibility. -

Annual Report 2008 Find out More About GSK Online…
Do more, feel better, live longer Grow Deliver Simplify Annual Report 2008 Find out more about GSK online… www.gsk.com Website GlaxoSmithKline’s website www.gsk.com gives additional information on the Group. Information made available on the website does not constitute part of this Annual Report. Notice regarding limitations on Director liability under English Law Under the UK Companies Act 2006, a safe harbour limits the liability of Directors in respect of statements in and omissions from the Report of the Directors contained on pages 12 to 98. Under English law the Directors would be liable to the company (but not to any third party) if the Report of the Directors contains errors as a result of recklessness or knowing misstatement or dishonest concealment of a material fact, but would not otherwise be liable. Report of the Directors Pages 12 to 98 inclusive consist of a Report of the Directors that has been drawn up and presented in accordance with and in reliance upon English company law and the liabilities of the Directors in connection with that report shall be subject to the limitations and restrictions provided by such law. Cautionary statement regarding forward-looking statements The Group’s reports filed with or furnished to the US Securities and Exchange Commission (SEC), including this document and written information released, or oral statements made, to the public in the future by or on behalf of the Group, may contain forward-looking statements. Forward-looking statements give the Group’s current expectations or forecasts of future events. A shareholder can identify these statements by the fact that they do not relate strictly to historical or current facts. -

The Clinical Biochemistry of 5'-Nucleotidase*
ANNALS OF CLINICAL AND LABORATORY SCIENCE, Vol. 20, No. 2 Copyright © 1990, Institute for Clinical Science, Inc. The Clinical Biochemistry of 5'-Nucleotidase* F. WILLIAM SUNDERMAN JR., M.D. Departments of Laboratory Medicine and Pharmacology, University of Connecticut Medical School, Farmington, CT 06032 ABSTRACT This review delineates the subcellular distribution, biochemical charac teristics, and metabolic functions of 5'-nucleotidase (5'NT), summarizes the analytical biochemistry of 5'NT, and assesses the clinical significance of5'NT determinations in body fluids, cells, and tissues. Salient aspects of the clinical biochemistry of 5'NT, discussed herein, are as follows: (A) Serum 5'NT activity is generally elevated in hepatobiliary diseases, espe cially with intrahepatic obstruction, but, unlike serum alkaline phospha tase, serum 5'NT activity is not increased in infancy, childhood, preg nancy, or osteoblastic disorders. (B) In cancer patients, elevated serum 5'NT activity does not always indicate hepatobiliary involvement; in some cases, 5'NT may be released into serum from the primary tumor or local metastases. (C) Genetic deficiency of erythrocyte pyrimidine 5'NT activity is a common cause of hereditary non-spherocytic hemolytic anemia. (D) Acquired deficiency of erythrocyte pyrimidine 5'NT activity occurs in patients with P-thalassemia and lead poisoning. (E) 5'NT activity is low in circulating monocytes, increases markedly upon their differentiation to tissue macrophages, and subsequently diminishes during macrophage activation. (F) Lymphocyte ecto-5'NT activity, a plasma membrane marker of cell maturation, is generally low in immunodeficiency states, and undergoes characteristic changes in patients with certain lymphomas and leukemias. Introduction ribonucleosides and inorganic phos phate. -

Poly (I)-Poly (C)/Oligo (Dt)-Cellulosei... Sidney Pestka, James Mcinnes
Contents Vol. 72, No. 10 October 1975 INFORMATION TO CONTRIBUTORS ..............................iv...........................I..................... i-i AUTfHOR INDEX ................................................................................................. 4190 Physical Sciences APPLIED Adiabatic evolution of plasma equilibrium (bifurcation/isolation/weak solution/generalized differential MATHEMATICS equation/Tokamak) ............................................ H. Grad, P.N. Hu, and D. C. Stevens 3789-3793 CHEMISTRY Theoretical studies of metal-phosphate interactions: Interaction of Li+, Na+, K+, Be++, Mg++, and Ca++ with H2PO4- and (CH30)2PO2-: Implications for nucleic acid solvation (metal binding/phos- phate complexes/molecular orbital theory) ................Dennis S. Marynick and Henry F. Schaefer III 3794-3798 Maximum-valence radii of transition metals (covalent radii/enneacovalence) ............... Linus Pauling 3799-3801 Model of protein folding: Inclusion of short-, medium-, and long-range interactions (mechanism offolding/ contact free energies/range of interactions/Monte Carlo) ........... Seiji Tanaka and HaroldA. Scheraga 3802-3806 Matrix method for fhActuations and noise in kinetic systems (cross correlation function/noise power spectrum matrix/relaxation matrix/biochemical reaction kinetics/muscle contraction) .Yi-der Chen 3807-3811 MATHEMATICS Point estimates for probability moments (approximate methods/finite differences/numerical methods/prob- abilities) .......................... - ;Emilio Rosenblueth 3812-3814 -

Phosphodiesterases Expression During Murine Cardiac Development
International Journal of Molecular Sciences Article Phosphodiesterases Expression during Murine Cardiac Development Thays Maria da Conceição Silva Carvalho 1,†, Silvia Cardarelli 1,†, Mauro Giorgi 2, Andrea Lenzi 3, Andrea M. Isidori 3 and Fabio Naro 1,* 1 Department of Anatomical, Histological, Forensic and Orthopedic Sciences, Sapienza University, 00161 Rome, Italy; [email protected] (T.M.d.C.S.C.); [email protected] (S.C.) 2 Department of Biology and Biotechnology “C. Darwin”, Sapienza University, 00185 Rome, Italy; [email protected] 3 Department of Experimental Medicine, Sapienza University, 00161 Rome, Italy; [email protected] (A.L.); [email protected] (A.M.I.) * Correspondence: [email protected] † These authors contributed equally to this work. Abstract: 30-50 cyclic nucleotide phosphodiesterases (PDEs) are a large family of enzymes playing a fundamental role in the control of intracellular levels of cAMP and cGMP. Emerging evidence suggested an important role of phosphodiesterases in heart formation, but little is known about the expression of phosphodiesterases during cardiac development. In the present study, the pattern of expression and enzymatic activity of phosphodiesterases was investigated at different stages of heart formation. C57BL/6 mice were mated and embryos were collected from 14.5 to 18.5 days of development. Data obtained by qRT-PCR and Western blot analysis showed that seven different isoforms are expressed during heart development, and PDE1C, PDE2A, PDE4D, PDE5A and PDE8A Citation: Carvalho, T.M.d.C.S.; are modulated from E14.5 to E18.5. In heart homogenates, the total cAMP and cGMP hydrolytic Cardarelli, S.; Giorgi, M.; Lenzi, A.; activity is constant at the evaluated times, and PDE4 accounts for the majority of the cAMP hydrolyz- Isidori, A.M.; Naro, F. -

Glaxosmithkline Annual Report 2010
Do more, feel better, live longer GlaxoSmithKline Annual Report 2010 Contents Business review P08–P57 Business review 2010 Performance overview 08 Research and development 10 Pipeline summary 12 Products, competition and intellectual property 14 Regulation 18 Manufacturing and supply 19 Business review World market 20 This discusses our financial and non-financial activities, GSK sales performance 21 resources, development and performance during 2010 Segment reviews 22 and outlines the factors, including the trends and the Responsible business 29 principal risks and uncertainties, which are likely to Financial review 2010 34 affect future development. Financial position and resources 41 Financial review 2009 47 Governance and remuneration Risk factors 53 This discusses our management structures and governance procedures. It also sets out the Governance and remuneration P58–P101 Governance and remuneration Governance and remuneration remuneration policies operated for our Directors and Our Board 58 Corporate Executive Team members. Our Corporate Executive Team 60 Governance and policy 64 Financial statements Dialogue with shareholders 69 The financial statements provide a summary of the Internal control framework 71 Group’s financial performance throughout 2010 and its Committee reports 74 position as at 31st December 2010. The consolidated Remuneration policy 84 financial statements are prepared in accordance with Director terms and conditions 91 IFRS as adopted by the European Union and also IFRS as Director and Senior Management remuneration 94 issued by the International Accounting Standards Board. Directors’ interests 96 Directors’ interests in contracts 101 Shareholder information This includes the full product development pipeline and discusses shareholder return in the form of dividends and share price movements. -

Oxidative Stress Responses in Escherichia Coli and Salmonella Typhimurium
MICROBIOLOGICAL REVIEWS, Dec. 1991, p. 561-585 Vol. 55, No. 4 0146-07 49/91/040561-25$02. 00/0 Copyright © 1991, American Society for Microbiology Oxidative Stress Responses in Escherichia coli and Salmonella typhimurium SPENCER B. FARR1* AND TOKIO KOGOMA2 Department of Molecular and Cellular Toxicology, Harvard School of Public Health, Boston, Massachusetts 02115, 1 and Departments ofCell Biology and Microbiology, University of New Mexico Medical Center, Albuquerque, New Mexico 87131 2 INTRODUCTION AND SCOPE OF REVIEW ............................................................................ 561 OXIDATIVE STRESS RESPONSES .........................................................................................562 Active Oxygen Species ......................................................................................................... 562 Reactivities .....••••.•.......................•............................•.........•.......................................... 562 Sources ......................................•................•.......•.•.•..................................••...••....•.......563 Oxidative Stress and Cellular Responses .........................................................•.......•..•••••....•...563 Peroxide stress response •................•.......•.............................................................•...........564 Su peroxide stress response ...............•..........•.•........•....••....••.....................••.•••••...••.•..•..•..•.564 The two oxidative stress responses are distinct .......................................................................564