SLC12A4 SLC12A6 TEP V1.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
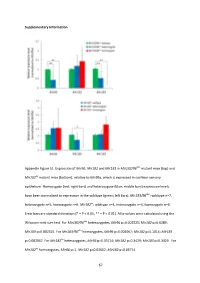
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

The Genetic Landscape of the Human Solute Carrier (SLC) Transporter Superfamily
Human Genetics (2019) 138:1359–1377 https://doi.org/10.1007/s00439-019-02081-x ORIGINAL INVESTIGATION The genetic landscape of the human solute carrier (SLC) transporter superfamily Lena Schaller1 · Volker M. Lauschke1 Received: 4 August 2019 / Accepted: 26 October 2019 / Published online: 2 November 2019 © The Author(s) 2019 Abstract The human solute carrier (SLC) superfamily of transporters is comprised of over 400 membrane-bound proteins, and plays essential roles in a multitude of physiological and pharmacological processes. In addition, perturbation of SLC transporter function underlies numerous human diseases, which renders SLC transporters attractive drug targets. Common genetic polymorphisms in SLC genes have been associated with inter-individual diferences in drug efcacy and toxicity. However, despite their tremendous clinical relevance, epidemiological data of these variants are mostly derived from heterogeneous cohorts of small sample size and the genetic SLC landscape beyond these common variants has not been comprehensively assessed. In this study, we analyzed Next-Generation Sequencing data from 141,456 individuals from seven major human populations to evaluate genetic variability, its functional consequences, and ethnogeographic patterns across the entire SLC superfamily of transporters. Importantly, of the 204,287 exonic single-nucleotide variants (SNVs) which we identifed, 99.8% were present in less than 1% of analyzed alleles. Comprehensive computational analyses using 13 partially orthogonal algorithms that predict the functional impact of genetic variations based on sequence information, evolutionary conserva- tion, structural considerations, and functional genomics data revealed that each individual genome harbors 29.7 variants with putative functional efects, of which rare variants account for 18%. Inter-ethnic variability was found to be extensive, and 83% of deleterious SLC variants were only identifed in a single population. -

Nephritis Responses in Murine and Human Lupus Analysis Defines
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology , 24 of which you can access for free at: 2012; 189:988-1001; Prepublished online 20 June from submission to initial decision 4 weeks from acceptance to publication Celine C. Berthier, Ramalingam Bethunaickan, Tania Gonzalez-Rivera, Viji Nair, Meera Ramanujam, Weijia Zhang, Erwin P. Bottinger, Stephan Segerer, Maja Lindenmeyer, Clemens D. Cohen, Anne Davidson and Matthias Kretzler 2012; doi: 10.4049/jimmunol.1103031 http://www.jimmunol.org/content/189/2/988 Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis J Immunol cites 60 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/189/2/988.full#ref-list-1 http://www.jimmunol.org/content/suppl/2012/06/20/jimmunol.110303 1.DC1 This article Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. The Journal of Immunology Cross-Species Transcriptional Network Analysis Defines Shared Inflammatory Responses in Murine and Human Lupus Nephritis Celine C. -

The Genetics of Bipolar Disorder
Molecular Psychiatry (2008) 13, 742–771 & 2008 Nature Publishing Group All rights reserved 1359-4184/08 $30.00 www.nature.com/mp FEATURE REVIEW The genetics of bipolar disorder: genome ‘hot regions,’ genes, new potential candidates and future directions A Serretti and L Mandelli Institute of Psychiatry, University of Bologna, Bologna, Italy Bipolar disorder (BP) is a complex disorder caused by a number of liability genes interacting with the environment. In recent years, a large number of linkage and association studies have been conducted producing an extremely large number of findings often not replicated or partially replicated. Further, results from linkage and association studies are not always easily comparable. Unfortunately, at present a comprehensive coverage of available evidence is still lacking. In the present paper, we summarized results obtained from both linkage and association studies in BP. Further, we indicated new potential interesting genes, located in genome ‘hot regions’ for BP and being expressed in the brain. We reviewed published studies on the subject till December 2007. We precisely localized regions where positive linkage has been found, by the NCBI Map viewer (http://www.ncbi.nlm.nih.gov/mapview/); further, we identified genes located in interesting areas and expressed in the brain, by the Entrez gene, Unigene databases (http://www.ncbi.nlm.nih.gov/entrez/) and Human Protein Reference Database (http://www.hprd.org); these genes could be of interest in future investigations. The review of association studies gave interesting results, as a number of genes seem to be definitively involved in BP, such as SLC6A4, TPH2, DRD4, SLC6A3, DAOA, DTNBP1, NRG1, DISC1 and BDNF. -

SLC12A6 Gene Solute Carrier Family 12 Member 6
SLC12A6 gene solute carrier family 12 member 6 Normal Function The SLC12A6 gene provides instructions for making a protein called a K-Cl cotransporter. This protein is involved in moving charged atoms (ions) of potassium (K) and chlorine (Cl) across the cell membrane. The positively charged potassium ions and negatively charged chlorine ions are moved together (cotransported), so that the charges inside and outside the cell membrane are unchanged (electroneutral). Electroneutral cotransport of ions across cell membranes is involved in many functions of the body. While the specific function of the K-Cl cotransporter produced from the SLC12A6 gene is unknown, it seems to be critical for the development and maintenance of nerve tissue. It may be involved in regulating the amounts of potassium, chlorine, or water in cells and intercellular spaces. The K-Cl cotransporter protein may also help regulate the activity of other proteins that are sensitive to ion concentrations. Health Conditions Related to Genetic Changes Andermann syndrome At least six SLC12A6 gene mutations have been identified in people with Andermann syndrome. Almost all affected individuals of French-Canadian descent have the same mutation in both copies of the SLC12A6 gene, in which the DNA building block ( nucleotide) guanine is deleted at position 2436 (written as 2436delG). This mutation is common in the populations of the Saguenay-Lac-St.-Jean and Charlevoix regions of northeastern Quebec. Most SLC12A6 gene mutations that cause Andermann syndrome result in a K-Cl cotransporter protein that is shortened and nonfunctional. The lack of functional protein produced from the SLC12A6 gene is believed to interfere with the development of the corpus callosum and maintenance of the nerves that transmit signals needed for movement and sensation, resulting in the signs and symptoms of Andermann syndrome. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Genetic Background of Ataxia in Children Younger Than 5 Years in Finland E444
Volume 6, Number 4, August 2020 Neurology.org/NG A peer-reviewed clinical and translational neurology open access journal ARTICLE Genetic background of ataxia in children younger than 5 years in Finland e444 ARTICLE Cerebral arteriopathy associated with heterozygous variants in the casitas B-lineage lymphoma gene e448 ARTICLE Somatic SLC35A2 mosaicism correlates with clinical fi ndings in epilepsy brain tissuee460 ARTICLE Synonymous variants associated with Alzheimer disease in multiplex families e450 Academy Officers Neurology® is a registered trademark of the American Academy of Neurology (registration valid in the United States). James C. Stevens, MD, FAAN, President Neurology® Genetics (eISSN 2376-7839) is an open access journal published Orly Avitzur, MD, MBA, FAAN, President Elect online for the American Academy of Neurology, 201 Chicago Avenue, Ann H. Tilton, MD, FAAN, Vice President Minneapolis, MN 55415, by Wolters Kluwer Health, Inc. at 14700 Citicorp Drive, Bldg. 3, Hagerstown, MD 21742. Business offices are located at Two Carlayne E. Jackson, MD, FAAN, Secretary Commerce Square, 2001 Market Street, Philadelphia, PA 19103. Production offices are located at 351 West Camden Street, Baltimore, MD 21201-2436. Janis M. Miyasaki, MD, MEd, FRCPC, FAAN, Treasurer © 2020 American Academy of Neurology. Ralph L. Sacco, MD, MS, FAAN, Past President Neurology® Genetics is an official journal of the American Academy of Neurology. Journal website: Neurology.org/ng, AAN website: AAN.com CEO, American Academy of Neurology Copyright and Permission Information: Please go to the journal website (www.neurology.org/ng) and click the Permissions tab for the relevant Mary E. Post, MBA, CAE article. Alternatively, send an email to [email protected]. -

Supporting Information
Supporting Information Table S1. List of confirmed SLC transporters represented in Canine GeneChip. SLC family Members detected Members not detected SLC1: The high affinity glutamate and neutral amino acid SLC1A1 SLC1A2, SLC1A3, SLC1A6 transporter family SLC2: The facilitative GLUT transporter family SLC2A1, SLC2A8 SLC2A3, SLC2A9 SLC3: The heavy subunits of the heteromeric amino acid SLC3A1 transporters SLC4: The bicarbonate transporter family SLC4A11 SLC4A4, SLC4A8 SLC5: The sodium glucose cotransporter family SLC5A6 SLC5A3, SLC5A10, SLC5A12 SLC6: The sodium- and chloride- dependent SLC6A6, SLC6A12 SLCA18 neurotransmitter transporter family SLC7: The cationic amino acid transporter/glycoprotein- NR associated family SLC8: The Na+/Ca2+ exchanger family SLC8A1 SLC9: The Na+/H+ exchanger family SLC9A1, SLC9A6, SLC9A9 SLC10: The sodium bile salt cotransport family SLC10A2 SLC11: The proton coupled metal ion transporter family NR SLC12: The electroneutral cation-Cl cotransporter family SLC12A3, SLC12A6, SLC12A8 SLC13: The human Na+-sulfate/carboxylate cotransporter SLC13A2 family SLC14: The urea transporter family NR SLC15: The proton oligopeptide cotransporter family SLC15A2, SLC15A4 SLC15A1 SLC16: The monocarboxylate transporter family SLC16A13 SLC16A4 SLC17: The vesicular glutamate transporter family SLC17A3, SLC17A7 SLC18: The vesicular amine transporter family NR SLC19: The folate/thiamine transporter family NR SLC20: The type III Na+-phosphate cotransporter family NR SLC21/SLCO: The organic anion transporting family SLC21A3, SLC21A8, -

Gene Expression Profiling of Transporters in the Solute Carrier and ATP-Binding Cassette Superfamilies in Human Eye Substructures
NIH Public Access Author Manuscript Mol Pharm. Author manuscript; available in PMC 2014 May 23. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Pharm. 2013 February 4; 10(2): 650–663. doi:10.1021/mp300429e. Gene Expression Profiling of Transporters in the Solute Carrier and ATP-Binding Cassette Superfamilies in Human Eye Substructures Amber Dahlin1,2,^, Ethan Geier1,^, Sophie L. Stocker1, Cheryl D. Cropp1, Elena Grigorenko3, Michele Bloomer4, Julie Siegenthaler5, Lu Xu1, Anthony S. Basile6, Diane D-S. Tang-Liu6, and Kathy Giacomini1,* 1Department of Bioengineering and Therapeutic Science, University of California, San Francisco, San Francisco, CA USA 3Life Technologies, Woburn, MA USA 4Department of Ophthalmology, University of California San Francisco, San Francisco, CA USA 5Department of Neurology, University of California San Francisco, San Francisco, CA USA 6Allergan, Inc. Irvine, CA USA Abstract The barrier epithelia of the cornea and retina control drug and nutrient access to various compartments of the human eye. While ocular transporters are likely to play a critical role in homeostasis and drug delivery, little is known about their expression, localization and function. In this study, the mRNA expression levels of 445 transporters, metabolic enzymes, transcription factors and nuclear receptors were profiled in five regions of the human eye: cornea, iris, ciliary body, choroid and retina. Through RNA expression profiling and immunohistochemistry, several transporters were identified as putative targets for drug transport in ocular tissues. Our analysis identified SLC22A7 (OAT2), a carrier for the anti-viral drug, acyclovir, in the corneal epithelium, in addition to ABCG2 (BCRP), an important xenobiotic efflux pump, in retinal nerve fibers and the retinal pigment epithelium. -

Table SI. Enriched Genes in the Upregulated Genes of the Recovery Group According to the GO Molecular Function Terms. A, Downreg
Table SI. Enriched genes in the upregulated genes of the recovery group according to the GO Molecular Function terms. A, Downregulated genes Adjusted Total Molecular Rank P‑value genes (n) Function Genes 1 <0.001 266 GO:0019899 Raf1 Timp1 Tbc1d8 Ube2g2 Ube2z enzyme binding Lonrf3 Tbc1d15 Rnf144a Ube2g1 Shc3 Rgcc Rnf19a Ube2j2 Rnf138 Atg13 Cks1b Ube2j1 Rnf19b Trib1 Trib3 Abtb2 Rnf125 Cdc42ep3 Nploc4 Cdc42ep4 Cdc42ep2 Rab11fip5 Arih2 Brms1 Tmem189 Mef2d Hspb1 Cdk9 Ksr1 Tnfaip3 Net1 Rnf180 Fgr Bhlhe41 Irs2 Ppp1r15a Asb4 Trim72 Zfp36 Sfn Xpo6 Fap Sox9 Mapk7 Itga3 Tubb5 Daxx Klf4 Stat3 Gab2 Myo9b Cstb Hmox1 Por Bcl2l1 Plin5 Chp1 Ube2i Sash1 Sqstm1 Rxra Slpi Sdc4 Tnfaip1 Cd40 Slc12a4 Map2k3 Ywhah Ppp1r12a Cry1 Plek Egfr Tnip1 Npc1l1 Rock2 Map2k6 Per1 Nfkbia Bdkrb2 Prkch Hif1a Golga5 Ripk1 Map3k1 Glud1 Nufip1 Clu Spry2 Hcls1 Ifnar2 Tuba1b Cdkn1a Sik1 Tmem173 Map3k2 Tnf Riok3 Ptpn2 Cep192 Smad2 Fas Jak2 Ankrd1 Rela Rps6ka4 Ankrd2 Rabgef1 Prkar1b Nop58 Casp8 Cflar Hdac4 Sele Nek2 Optn Nek6 Lcn2 Stom Traf6 Spred1 Nop56 Src Ccnl1 Ptpn22 Il6ra Pip5k1a F3 Bcl10 3110043O21Rik Tnfrsf1b Slc2a1 Sfpq Rpa2 Errfi1 Mad2l2 Tbc1d14 Uchl1 Glmn Scarb2 Ulk1 Ung Rad18 Mef2a Ctsc Ipo5 Mvp Kctd13 Msn Eif4ebp1 Casp3 Smad1 Ubash3b Ets1 Tirap Smad3 Tgfbr2 Ptgs2 Prr5l Micall1 Cnppd1 Map2k4 Tnks1bp1 Ppp1r32 Prdm4 Midn Ibtk Rusc2 Fmnl2 Ptpn23 Sh3bp4 Nop14 Kdm1a Serpine1 Gch1 Inf2 Csf3 Snx10 Txnip Egr1 Ranbp9 Akap12 Rab3gap2 Ddx58 Bcor Rabggta Pik3r1 Pkp2 Usp22 Shc1 Ptpn11 Fzd5 Cxcr4 Plaur Bag5 Maml1 Camk2n2 Taf7 Ywhag Ezr Jun Camk2d Parp4 Nod2 Ptafr Hmga2 Zfp746 Ptk2b Flot1 -

Chloride Homeostasis in Neurons with Special Emphasis on the Olivocerebellar System: Differential Roles for Transporters and Channels
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Erasmus University Digital Repository REVIEW published: 01 May 2018 doi: 10.3389/fncel.2018.00101 Chloride Homeostasis in Neurons With Special Emphasis on the Olivocerebellar System: Differential Roles for Transporters and Channels Negah Rahmati 1*†, Freek E. Hoebeek 1,2, Saša Peter 1 and Chris I. De Zeeuw 1,3 1 Department of Neuroscience, Erasmus Medical Center, Rotterdam, Netherlands, 2 NIDOD Institute, Wilhelmina Children’s Hospital, University Medical Center Utrecht and Brain Center Rudolf Magnus, Utrecht, Netherlands, 3 Netherlands Institute for Neuroscience, Royal Dutch Academy for Arts and Sciences, Amsterdam, Netherlands The intraneuronal ionic composition is an important determinant of brain functioning. There is growing evidence that aberrant homeostasis of the intracellular concentration of − − + 2+ Cl ([Cl ]i) evokes, in addition to that of Na and Ca , robust impairments of neuronal excitability and neurotransmission and thereby neurological conditions. More specifically, − Edited by: understanding the mechanisms underlying regulation of [Cl ]i is crucial for deciphering Jonathan Mapelli, the variability in GABAergic and glycinergic signaling of neurons, in both health and University of Modena and Reggio − Emilia, Italy disease. The homeostatic level of [Cl ]i is determined by various regulatory mechanisms, − Reviewed by: including those mediated by plasma membrane Cl channels and transporters. This Mauricio Di Fulvio, review focuses -

RNA-Seq Reveals Conservation of Function Among the Yolk Sacs Of
RNA-seq reveals conservation of function among the PNAS PLUS yolk sacs of human, mouse, and chicken Tereza Cindrova-Daviesa, Eric Jauniauxb, Michael G. Elliota,c, Sungsam Gongd,e, Graham J. Burtona,1, and D. Stephen Charnock-Jonesa,d,e,1,2 aCentre for Trophoblast Research, Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, CB2 3EG, United Kingdom; bElizabeth Garret Anderson Institute for Women’s Health, Faculty of Population Health Sciences, University College London, London, WC1E 6BT, United Kingdom; cSt. John’s College, University of Cambridge, Cambridge, CB2 1TP, United Kingdom; dDepartment of Obstetrics and Gynaecology, University of Cambridge, Cambridge, CB2 0SW, United Kingdom; and eNational Institute for Health Research, Cambridge Comprehensive Biomedical Research Centre, Cambridge, CB2 0QQ, United Kingdom Edited by R. Michael Roberts, University of Missouri-Columbia, Columbia, MO, and approved May 5, 2017 (received for review February 14, 2017) The yolk sac is phylogenetically the oldest of the extraembryonic yolk sac plays a critical role during organogenesis (3–5, 8–10), membranes. The human embryo retains a yolk sac, which goes there are limited data to support this claim. Obtaining experi- through primary and secondary phases of development, but its mental data for the human is impossible for ethical reasons, and importance is controversial. Although it is known to synthesize thus we adopted an alternative strategy. Here, we report RNA proteins, its transport functions are widely considered vestigial. sequencing (RNA-seq) data derived from human and murine yolk Here, we report RNA-sequencing (RNA-seq) data for the human sacs and compare them with published data from the yolk sac of and murine yolk sacs and compare those data with data for the the chicken.