Parental-To-Embryo Switch of Chromosome Organization in Early
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -
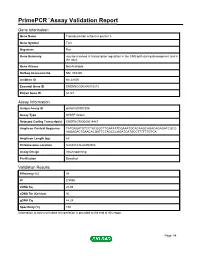
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Transducin-like enhancer protein 3 Gene Symbol Tle3 Organism Rat Gene Summary may be involved in transcription regulation in the CNS both during development and in the adult Gene Aliases Not Available RefSeq Accession No. NM_053400 UniGene ID Rn.24106 Ensembl Gene ID ENSRNOG00000013013 Entrez Gene ID 84424 Assay Information Unique Assay ID qRnoCID0007306 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000018467 Amplicon Context Sequence TATGAGATGTCCTACGGCTTGAATATCGAAATGCACAAGCAGACAGAGATCGCG AAGAGACTGAACACGATTCTAGCCCAGATCATGCCTTTTTTGTCA Amplicon Length (bp) 69 Chromosome Location 8:66041636-66059306 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9996 cDNA Cq 20.98 cDNA Tm (Celsius) 80 gDNA Cq 44.24 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Tle3, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Gene Expression Profiling of Human Sarcomas: Insights Into Sarcoma Biology
Research Article Gene Expression Profiling of Human Sarcomas: Insights into Sarcoma Biology Kristin Baird,1 Sean Davis,1 Cristina R. Antonescu,3 Ursula L. Harper,1 Robert L. Walker,1 Yidong Chen,1 Arthur A. Glatfelter,1 Paul H. Duray,2 and Paul S. Meltzer1 1Cancer Genetics Branch, National Human Genome Research Institute; 2Laboratory of Pathology, National Cancer Institute, NIH, Bethesda, Maryland; 3Department of Pathology, Memorial Sloan-Kettering Cancer Center, New York, New York Abstract changes. This latter group occurs more frequently in older adults and includes several types of sarcomas that lack known disease- Sarcomas are a biologically complex group of tumors of specific chromosome translocations or mutations, but may contain mesenchymal origin. By using gene expression microarray mutations in RB1, CDKN2A, and TP53. Investigation of these and analysis, we aimed to find clues into the cellular differenti- other aspects of sarcoma biology have provided insights into ation and oncogenic pathways active in these tumors as well broadly relevant fundamental mechanisms of oncogenesis (for as potential biomarkers and therapeutic targets. We exam- review, see ref. 1) and may have profound implications in the ined 181 tumors representing 16 classes of human bone and development of therapeutic intervention. This is exemplified by the soft tissue sarcomas on a 12,601-feature cDNA microarray. successful treatment of gastrointestinal stromal tumors, which Remarkably, 2,766 probes differentially expressed across this frequently have activating mutations of KIT, with the tyrosine sample set clearly delineated the various tumor classes. kinase inhibitor, imatinib mesylate. Although genetic alterations, Several genes of potential biological and therapeutic interest particularly fusion genes arising from translocations, have been were associated with each sarcoma type, including specific identified in many sarcomas, the function of the fusion gene tyrosine kinases, transcription factors, and homeobox genes. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Newly Identified Gon4l/Udu-Interacting Proteins
www.nature.com/scientificreports OPEN Newly identifed Gon4l/ Udu‑interacting proteins implicate novel functions Su‑Mei Tsai1, Kuo‑Chang Chu1 & Yun‑Jin Jiang1,2,3,4,5* Mutations of the Gon4l/udu gene in diferent organisms give rise to diverse phenotypes. Although the efects of Gon4l/Udu in transcriptional regulation have been demonstrated, they cannot solely explain the observed characteristics among species. To further understand the function of Gon4l/Udu, we used yeast two‑hybrid (Y2H) screening to identify interacting proteins in zebrafsh and mouse systems, confrmed the interactions by co‑immunoprecipitation assay, and found four novel Gon4l‑interacting proteins: BRCA1 associated protein‑1 (Bap1), DNA methyltransferase 1 (Dnmt1), Tho complex 1 (Thoc1, also known as Tho1 or HPR1), and Cryptochrome circadian regulator 3a (Cry3a). Furthermore, all known Gon4l/Udu‑interacting proteins—as found in this study, in previous reports, and in online resources—were investigated by Phenotype Enrichment Analysis. The most enriched phenotypes identifed include increased embryonic tissue cell apoptosis, embryonic lethality, increased T cell derived lymphoma incidence, decreased cell proliferation, chromosome instability, and abnormal dopamine level, characteristics that largely resemble those observed in reported Gon4l/udu mutant animals. Similar to the expression pattern of udu, those of bap1, dnmt1, thoc1, and cry3a are also found in the brain region and other tissues. Thus, these fndings indicate novel mechanisms of Gon4l/ Udu in regulating CpG methylation, histone expression/modifcation, DNA repair/genomic stability, and RNA binding/processing/export. Gon4l is a nuclear protein conserved among species. Animal models from invertebrates to vertebrates have shown that the protein Gon4-like (Gon4l) is essential for regulating cell proliferation and diferentiation. -

Identification of Protein Features Encoded by Alternative Exons Using Exon Ontology
Downloaded from genome.cshlp.org on October 2, 2021 - Published by Cold Spring Harbor Laboratory Press Resource Identification of protein features encoded by alternative exons using Exon Ontology Léon-Charles Tranchevent,1 Fabien Aubé,1 Louis Dulaurier,1 Clara Benoit-Pilven,1 Amandine Rey,1 Arnaud Poret,1 Emilie Chautard,2 Hussein Mortada,1 François-Olivier Desmet,1 Fatima Zahra Chakrama,1 Maira Alejandra Moreno-Garcia,1 Evelyne Goillot,3 Stéphane Janczarski,1 Franck Mortreux,1 Cyril F. Bourgeois,1,4 and Didier Auboeuf1,4 1Université Lyon 1, ENS de Lyon, CNRS UMR 5239, INSERM U1210, Laboratory of Biology and Modelling of the Cell, F-69007, Lyon, France; 2Laboratoire de Biométrie et Biologie Évolutive, Université Lyon 1, UMR CNRS 5558, INRIA Erable, Villeurbanne, F-69622, France; 3Institut NeuroMyoGène, CNRS UMR 5310, INSERM U1217, Université Lyon 1, Lyon, F-69007 France Transcriptomic genome-wide analyses demonstrate massive variation of alternative splicing in many physiological and pathological situations. One major challenge is now to establish the biological contribution of alternative splicing var- iation in physiological- or pathological-associated cellular phenotypes. Toward this end, we developed a computational approach, named Exon Ontology, based on terms corresponding to well-characterized protein features organized in an ontology tree. Exon Ontology is conceptually similar to Gene Ontology-based approaches but focuses on exon-encod- ed protein features instead of gene level functional annotations. Exon Ontology describes the protein features encoded by a selected list of exons and looks for potential Exon Ontology term enrichment. By applying this strategy to exons that are differentially spliced between epithelial and mesenchymal cells and after extensive experimental validation, we demonstrate that Exon Ontology provides support to discover specific protein features regulated by alternative splic- ing. -

ID AKI Vs Control Fold Change P Value Symbol Entrez Gene Name *In
ID AKI vs control P value Symbol Entrez Gene Name *In case of multiple probesets per gene, one with the highest fold change was selected. Fold Change 208083_s_at 7.88 0.000932 ITGB6 integrin, beta 6 202376_at 6.12 0.000518 SERPINA3 serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3 1553575_at 5.62 0.0033 MT-ND6 NADH dehydrogenase, subunit 6 (complex I) 212768_s_at 5.50 0.000896 OLFM4 olfactomedin 4 206157_at 5.26 0.00177 PTX3 pentraxin 3, long 212531_at 4.26 0.00405 LCN2 lipocalin 2 215646_s_at 4.13 0.00408 VCAN versican 202018_s_at 4.12 0.0318 LTF lactotransferrin 203021_at 4.05 0.0129 SLPI secretory leukocyte peptidase inhibitor 222486_s_at 4.03 0.000329 ADAMTS1 ADAM metallopeptidase with thrombospondin type 1 motif, 1 1552439_s_at 3.82 0.000714 MEGF11 multiple EGF-like-domains 11 210602_s_at 3.74 0.000408 CDH6 cadherin 6, type 2, K-cadherin (fetal kidney) 229947_at 3.62 0.00843 PI15 peptidase inhibitor 15 204006_s_at 3.39 0.00241 FCGR3A Fc fragment of IgG, low affinity IIIa, receptor (CD16a) 202238_s_at 3.29 0.00492 NNMT nicotinamide N-methyltransferase 202917_s_at 3.20 0.00369 S100A8 S100 calcium binding protein A8 215223_s_at 3.17 0.000516 SOD2 superoxide dismutase 2, mitochondrial 204627_s_at 3.04 0.00619 ITGB3 integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) 223217_s_at 2.99 0.00397 NFKBIZ nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta 231067_s_at 2.97 0.00681 AKAP12 A kinase (PRKA) anchor protein 12 224917_at 2.94 0.00256 VMP1/ mir-21likely ortholog -

Towards Personalized Medicine in Psychiatry: Focus on Suicide
TOWARDS PERSONALIZED MEDICINE IN PSYCHIATRY: FOCUS ON SUICIDE Daniel F. Levey Submitted to the faculty of the University Graduate School in partial fulfillment of the requirements for the degree Doctor of Philosophy in the Program of Medical Neuroscience, Indiana University April 2017 ii Accepted by the Graduate Faculty, Indiana University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy. Andrew J. Saykin, Psy. D. - Chair ___________________________ Alan F. Breier, M.D. Doctoral Committee Gerry S. Oxford, Ph.D. December 13, 2016 Anantha Shekhar, M.D., Ph.D. Alexander B. Niculescu III, M.D., Ph.D. iii Dedication This work is dedicated to all those who suffer, whether their pain is physical or psychological. iv Acknowledgements The work I have done over the last several years would not have been possible without the contributions of many people. I first need to thank my terrific mentor and PI, Dr. Alexander Niculescu. He has continuously given me advice and opportunities over the years even as he has suffered through my many mistakes, and I greatly appreciate his patience. The incredible passion he brings to his work every single day has been inspirational. It has been an at times painful but often exhilarating 5 years. I need to thank Helen Le-Niculescu for being a wonderful colleague and mentor. I learned a lot about organization and presentation working alongside her, and her tireless work ethic was an excellent example for a new graduate student. I had the pleasure of working with a number of great people over the years. Mikias Ayalew showed me the ropes of the lab and began my understanding of the power of algorithms. -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. -

Loss of TLE3 Promotes the Mitochondrial Program in Beige Adipocytes and Improves Glucose Metabolism
Downloaded from genesdev.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Loss of TLE3 promotes the mitochondrial program in beige adipocytes and improves glucose metabolism Stephanie Pearson,1 Anne Loft,2 Prashant Rahbhandari,3 Judith Simcox,1 Sanghoon Lee,1 Peter Tontonoz,3 Susanne Mandrup,2 and Claudio J. Villanueva1 1Department of Biochemistry, University of Utah School of Medicine, Salt Lake City, Utah 84112, USA; 2Department of Biochemistry and Molecular Biology, University of Southern Denmark, 5230 Odense M, Denmark; 3Department of Pathology and Laboratory Medicine, University of California at Los Angeles, Los Angeles, California 90095, USA Prolonged cold exposure stimulates the recruitment of beige adipocytes within white adipose tissue. Beige adipo- cytes depend on mitochondrial oxidative phosphorylation to drive thermogenesis. The transcriptional mechanisms that promote remodeling in adipose tissue during the cold are not well understood. Here we demonstrate that the transcriptional coregulator transducin-like enhancer of split 3 (TLE3) inhibits mitochondrial gene expression in beige adipocytes. Conditional deletion of TLE3 in adipocytes promotes mitochondrial oxidative metabolism and increases energy expenditure, thereby improving glucose control. Using chromatin immunoprecipitation and deep sequencing, we found that TLE3 occupies distal enhancers in proximity to nuclear-encoded mitochondrial genes and that many of these binding sites are also enriched for early B-cell factor (EBF) transcription factors. TLE3 interacts with EBF2 and blocks its ability to promote the thermogenic transcriptional program. Collectively, these studies demonstrate that TLE3 regulates thermogenic gene expression in beige adipocytes through inhibition of EBF2 transcriptional activity. Inhibition of TLE3 may provide a novel therapeutic approach for obesity and diabetes. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ......................................................