RAB23 Coordinates Early Osteogenesis by Repressing FGF10
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanical Forces Induce an Asthma Gene Signature in Healthy Airway Epithelial Cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T
www.nature.com/scientificreports OPEN Mechanical forces induce an asthma gene signature in healthy airway epithelial cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T. Kho4, Kelan Tantisira1, Marc Santolini 1,5, Feixiong Cheng6,7,8, Jennifer A. Mitchel3, Maureen McGill3, Michael J. O’Sullivan3, Margherita De Marzio1,3, Amitabh Sharma1, Scott H. Randell9, Jefrey M. Drazen3, Jefrey J. Fredberg3 & Scott T. Weiss1,3* Bronchospasm compresses the bronchial epithelium, and this compressive stress has been implicated in asthma pathogenesis. However, the molecular mechanisms by which this compressive stress alters pathways relevant to disease are not well understood. Using air-liquid interface cultures of primary human bronchial epithelial cells derived from non-asthmatic donors and asthmatic donors, we applied a compressive stress and then used a network approach to map resulting changes in the molecular interactome. In cells from non-asthmatic donors, compression by itself was sufcient to induce infammatory, late repair, and fbrotic pathways. Remarkably, this molecular profle of non-asthmatic cells after compression recapitulated the profle of asthmatic cells before compression. Together, these results show that even in the absence of any infammatory stimulus, mechanical compression alone is sufcient to induce an asthma-like molecular signature. Bronchial epithelial cells (BECs) form a physical barrier that protects pulmonary airways from inhaled irritants and invading pathogens1,2. Moreover, environmental stimuli such as allergens, pollutants and viruses can induce constriction of the airways3 and thereby expose the bronchial epithelium to compressive mechanical stress. In BECs, this compressive stress induces structural, biophysical, as well as molecular changes4,5, that interact with nearby mesenchyme6 to cause epithelial layer unjamming1, shedding of soluble factors, production of matrix proteins, and activation matrix modifying enzymes, which then act to coordinate infammatory and remodeling processes4,7–10. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -
Type of the Paper (Article
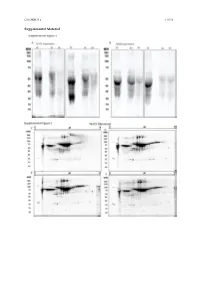
Cells 2020, 9, x 1 of 19 Supplemental Material Cells 2020, 9, x 2 of 19 Figure S1. Secretome enrichment: protocol optimization. 1D SDS-PAGE documentation of washing steps: Culture medium was substituted with FCS-free medium, which was changed every 2 h. The supernatants were then collected, and the proteins isolated and separated in 1D SDS-PAGE ((A) TK173 and (B) TK188). Proteins were stained with Flamingo fluorescent gel stain. Two-dimensional pattern of the proteins isolated from supernatant of TK173, (C) 2 h, (D) 4 h, (E) 6 h, and (F) 8 h after changing to FCS-free medium. (G) Cell secretome collected 24 h after elimination of the contaminating FCS-proteins with different washing steps. Proteins were stained with Flamingo fluorescent gel stain. Cells 2020, 9, x 3 of 19 Figure S2. 2-DE reference maps of secretomes; 150 μg proteins were loaded on an 11 cm IPG strip with a linear pH gradient PI 5–8 for IEF; 12% SDS-polyacrylamide gels were used for the second dimension. Proteins were stained with Flamingo fluorescent gel stain. Identified spots were assigned a number corresponding to that in their table. 2-DE maps from secretome of (A) TK173 control and (B) TGFβ1- treated ones. The 2-DE patterns revealed an alteration of secretome in stimulated TK173. Secretome patterns from TK173 treated with (C) ANG II and (D) PDGF. Cells 2020, 9, x 4 of 19 A Figure S3. Classification of the differentially expressed proteins upon ANG II, TGFβ1, or PDGF treatment in TK173. (A) Bar charts of the cellular component analyzed by STRAP biological function analysis in which the identified proteins from all treatments in both cell types are involved. -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

Origin, Characterization and Roles of Matrix Vesicles in Physiological and Pathological Mineralization
Origin, characterization and roles of matrix vesicles in physiological and pathological mineralization. Cyril Thouverey To cite this version: Cyril Thouverey. Origin, characterization and roles of matrix vesicles in physiological and pathological mineralization.. Life Sciences [q-bio]. Université Claude Bernard - Lyon I, 2008. English. tel- 00304214 HAL Id: tel-00304214 https://tel.archives-ouvertes.fr/tel-00304214 Submitted on 22 Jul 2008 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. N° d’ordre: 95-2008 Année 2008 THESE Présentée devant Nencki Institute of Experimental Biology, Polish Academy of Sciences Université Claude Bernard Lyon 1, Pour l’obtention Du DIPLOME DE DOCTORAT (Arrêté du 7 août 2006 et arrêté du 6 janvier 2005) Présentée et soutenue publiquement le 20 juin 2008 Par Cyril Thouverey Origin, characterization and roles of matrix vesicles in physiological and pathological mineralization Directeurs de thèse: Pr René Buchet et Pr Sławomir Pikuła JURY: Mme Françoise Bleicher Mme Joanna Bandorowicz-Pikuła Mme Martime Cohen-Solal Mr René Buchet Mr Sławomir Pikuła Mr Aleksander Sikorski UNIVERSITE CLAUDE BERNARD - LYON I Président de l’Université M. le Professeur L. COLLET Vice-Président du Conseil Scientifique M. -

Expression of Primary Cilia-Related Genes in Developing Mouse Gonads RAFAL P
Int. J. Dev. Biol. 63: 615-621 (2019) https://doi.org/10.1387/ijdb.190049rp www.intjdevbiol.com Expression of primary cilia-related genes in developing mouse gonads RAFAL P. PIPREK*,1, DAGMARA PODKOWA1, MALGORZATA KLOC2,3,4 and JACEK Z. KUBIAK5,6 1Department of Comparative Anatomy, Institute of Zoology and Biomedical Research, Jagiellonian University, Krakow, Poland, 2The Houston Methodist Research Institute, Houston, TX, USA, 3Department of Surgery, The Houston Methodist Hospital, Houston TX, USA, 4University of Texas, MD Anderson Cancer Center, Houston TX, USA, 5Univ Rennes, CNRS, Institute of Genetics and Development of Rennes, UMR 6290, Cell Cycle Group, Faculty of Medicine, Rennes, France and 6Laboratory of Regenerative Medicine and Cell Biology, Military Institute of Hygiene and Epidemiology (WIHE), Warsaw, Poland ABSTRACT Mechanisms governing differentiation of the bipotential gonad into the testes or ovaries are complex and still vague. The primary cilium is an organelle involved in cell signaling, which controls the development of many organs, but the role of primary cilium in the sex determination and sexual differentiation of gonads is completely unknown. Here we studied the expression of genes involved in primary cilium formation and functioning in fetal mouse gonads, before, during and after sexual differentiation. We studied the expression of 175 primary cilia-related genes using microarray technique. 144 of these genes were ubiquitously expressed in all studied cell types with no significant differences in expression level. Such a high level of expression of primary cilia-related genes in developing mouse gonads suggests that the primary cilia and/or primary cilia-related genes are important for the development of both somatic and germline component of the gonads. -

Functional Study of Mir-27A in Human Hepatic Stellate Cells by Proteomic Analysis: Comprehensive View and a Role in Myogenic Tans-Differentiation
Functional Study of miR-27a in Human Hepatic Stellate Cells by Proteomic Analysis: Comprehensive View and a Role in Myogenic Tans-Differentiation Yuhua Ji1, Jinsheng Zhang2, Wenwen Wang3, Juling Ji3* 1 Key Laboratory of Neuroregeneration, Nantong University, Nanton, China, 2 Department of Pathology, Shanghai Medical College, Fudan University, Shanghai, PR China, 3 Department of Pathology, Medical School of Nantong University, Nantong, PR China Abstract We previous reported that miR-27a regulates lipid metabolism and cell proliferation during hepatic stellate cells (HSCs) activation. To further explore the biological function and underlying mechanisms of miR-27a in HSCs, global protein expression affected by overexpression of miR-27a in HSCs was analyzed by a cleavable isotope-coded affinity tags (cICAT) based comparative proteomic approach. In the present study, 1267 non-redundant proteins were identified with unique accession numbers (score $1.3, i.e. confidence $95%), among which 1171 were quantified and 149 proteins (12.72%) were differentially expressed with a differential expression ratio of 1.5. We found that up-regulated proteins by miR-27a mainly participate in cell proliferation and myogenesis, while down-regulated proteins were the key enzymes involved in de novo lipid synthesis. The expression of a group of six miR-27a regulated proteins was validated and the function of one miR-27a regulated protein was further validated. The results not only delineated the underlying mechanism of miR-27a in modulating fat metabolism and cell proliferation, but also revealed a novel role of miR-27a in promoting myogenic tans- differentiation during HSCs activation. This study also exemplified proteomics strategy as a powerful tool for the functional study of miRNA. -

ARL13B, PDE6D, and CEP164 Form a Functional Network for INPP5E Ciliary Targeting
ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting Melissa C. Humberta,b, Katie Weihbrechta,b, Charles C. Searbyb,c, Yalan Lid, Robert M. Poped, Val C. Sheffieldb,c, and Seongjin Seoa,1 aDepartment of Ophthalmology and Visual Sciences, bDepartment of Pediatrics, cHoward Hughes Medical Institute, and dProteomics Facility, University of Iowa, Iowa City, IA 52242 Edited by Kathryn V. Anderson, Sloan-Kettering Institute, New York, NY, and approved October 19, 2012 (received for review June 28, 2012) Mutations affecting ciliary components cause a series of related polydactyly, skeletal defects, cleft palate, and cerebral develop- genetic disorders in humans, including nephronophthisis (NPHP), mental defects (11). Inactivation of Inpp5e in adult mice results in Joubert syndrome (JBTS), Meckel-Gruber syndrome (MKS), and Bar- obesity and photoreceptor degeneration. Interestingly, many pro- det-Biedl syndrome (BBS), which are collectively termed “ciliopa- teins that localize to cilia, including INPP5E, RPGR, PDE6 α and thies.” Recent protein–protein interaction studies combined with β subunits, GRK1 (Rhodopsin kinase), and GNGT1 (Transducin γ genetic analyses revealed that ciliopathy-related proteins form sev- chain), are prenylated (either farnesylated or geranylgeranylated), eral functional networks/modules that build and maintain the pri- and mutations in these genes or genes involved in their prenylation mary cilium. However, the precise function of many ciliopathy- (e.g., AIPL1 and RCE1) lead to photoreceptor -

Epstein-Barr Virus Induction of the Hedgehog Signalling Pathway Imposes a Stem Cell-Like Phenotype on Human Epithelial Cells –
Epstein-Barr Virus Induction of the Hedgehog Signalling Pathway Imposes a Stem Cell-Like Phenotype on Human Epithelial Cells – Implications for the Pathogenesis of Nasopharyngeal Carcinoma by REBECCA J. PORT A thesis submitted to the University of Birmingham for the degree of DOCTOR OF PHILOSOPHY School of Cancer Sciences College of Medical and Dental Sciences University of Birmingham September 2013 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Nasopharyngeal carcinoma (NPC) is endemic in Southern China and South East Asia, causally linked to Epstein-Barr virus (EBV) infection, and frequently shows dysregulation in a number of stem cell maintenance signalling pathways. This thesis has endeavoured to investigate the status of one of these pathways; the Hedgehog (HH) signalling pathway, in NPC tumours, and reveals the novel finding that EBV is able to active the HH signalling pathway through autocrine induction of the SHH ligand in the C666.1 authentic EBV-positive NPC-derived cell line and latently infected epithelial carcinoma cell lines. This study demonstrates that constitutive engagement of the HH pathway in EBV-infected epithelial cells in vitro induces the expression of a number of stemness-associated genes and imposes stem-like characteristics. -

Sonic Hedgehog Regulates Integrin Activity, Cadherin Contacts, and Cell Polarity to Orchestrate Neural Tube Morphogenesis
12506 • The Journal of Neuroscience, October 7, 2009 • 29(40):12506–12520 Development/Plasticity/Repair Sonic Hedgehog Regulates Integrin Activity, Cadherin Contacts, and Cell Polarity to Orchestrate Neural Tube Morphogenesis Claire Fournier-Thibault,1,2 Ce´drine Blavet,1,2 Artem Jarov,1,2 Fernanda Bajanca,3,4 So´lveig Thorsteinsdo´ttir,3,4 and Jean-Loup Duband1,2 1Universite´ Pierre et Marie Curie–Paris 6, Laboratoire de Biologie du De´veloppement, and 2Centre National de la Recherche Scientifique, Laboratoire de Biologie du De´veloppement, 75005 Paris, France, 3Departamento de Biologia Animal, Centro de Biologia Ambiental, Faculdade de Cieˆncias, Universidade de Lisboa, 1749-016 Lisboa, Portugal, and 4Instituto Gulbenkian de Cieˆncia, 2781-901 Oeiras, Portugal In vertebrates, the embryonic nervous system is shaped and patterned by a series of temporally and spatially regulated cell divisions, cell specifications, and cell adhesions and movements. Morphogens of the Hedgehog, Wnt, and bone morphogenetic protein families have been shown to play a crucial role in the control of cell division and specification in the trunk neural tube, but their possible implication in theregulationofadhesiveeventshasbeenpoorlydocumented.Inthepresentstudy,wedemonstratethatSonichedgehogregulatesneural epithelialcelladhesionandpolaritythroughregulationofintegrinactivity,cadherincell–cellcontact,andcellpolaritygenesinimmature neural epithelial cells before the specification of neuronal cells. We propose that Sonic hedgehog orchestrates neural tube morphogenesis -

Expression of Primary Cilia-Related Genes in Developing Mouse Gonads R.P
Expression of primary cilia-related genes in developing mouse gonads R.P. Piprek, D. Podkowa, M. Kloc, Jacek Kubiak To cite this version: R.P. Piprek, D. Podkowa, M. Kloc, Jacek Kubiak. Expression of primary cilia-related genes in de- veloping mouse gonads. International Journal of Developmental Biology, University of the Basque Country Press, 2019, 63 (11-12), pp.615-621. 10.1387/ijdb.190049rp. hal-02569583 HAL Id: hal-02569583 https://hal-univ-rennes1.archives-ouvertes.fr/hal-02569583 Submitted on 31 Aug 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Expression of primary cilia markers in developing mouse gonads 2 3 Rafal P. Piprek1*, Dagmara Podkowa1, Malgorzata Kloc2,3,4, Jacek Z. Kubiak5,6 4 5 1Department of Comparative Anatomy, Institute of Zoology and Biomedical Research, 6 Jagiellonian University, Krakow, Poland 7 2The Houston Methodist Research Institute, Houston, TX, USA 8 3Department of Surgery, The Houston Methodist Hospital, Houston TX, USA 9 4University of Texas, MD Anderson Cancer Center, Houston TX, USA 10 5Univ Rennes, CNRS, Institute of Genetics and Development of Rennes, UMR 6290, Cell 11 Cycle Group, Faculty of Medicine, F-35000 Rennes, France 12 6Laboratory of Regenerative Medicine and Cell Biology, Military Institute of Hygiene and 13 Epidemiology (WIHE), Warsaw, Poland 14 15 Running title: Primary cilia markers in developing mouse gonads 16 Key words: gonad development; sex determination; ovary; testis; primary cilia 17 18 Corresponding author: 19 Rafal P. -

Drosophila Rab23 Is Involved in the Regulation of the Number and Planar Polarization of the Adult Cuticular Hairs
Copyright Ó 2010 by the Genetics Society of America DOI: 10.1534/genetics.109.112060 Drosophila Rab23 Is Involved in the Regulation of the Number and Planar Polarization of the Adult Cuticular Hairs Csilla Pataki,*,1 Tama´s Matusek,*,1 E´ va Kurucz,* Istva´n Ando´,* Andreas Jenny† and Jo´zsef Miha´ly*,2 *Institute of Genetics, Biological Research Center, Hungarian Academy of Sciences, Szeged 6726, Hungary and †Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, New York 10461 Manuscript received November 13, 2009 Accepted for publication January 27, 2010 ABSTRACT The planar coordination of cellular polarization is an important, yet not well-understood aspect of animal development. In a screen for genes regulating planar cell polarization in Drosophila, we identified Rab23, encoding a putative vesicular trafficking protein. Mutations in the Drosophila Rab23 ortholog result in abnormal trichome orientation and the formation of multiple hairs on the wing, leg, and abdomen. We show that Rab23 is required for hexagonal packing of the wing cells. We found that Rab23 is able to associate with the proximally accumulated Prickle protein, although Rab23 itself does not seem to display a polarized subcellular distribution in wing cells, and it appears to play a relatively subtle role in cortical polarization of the polarity proteins. The absence of Rab23 leads to increased actin accumulation in the subapical region of the pupal wing cells that fail to restrict prehair initiation to a single site. Rab23 acts as a dominant enhancer of the weak multiple hair phenotype exhibited by the core polarity mutations, whereas the Rab23 homozygous mutant phenotype is sensitive to the gene dose of the planar polarity effector genes.