Epstein-Barr Virus Induction of the Hedgehog Signalling Pathway Imposes a Stem Cell-Like Phenotype on Human Epithelial Cells –
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mechanical Forces Induce an Asthma Gene Signature in Healthy Airway Epithelial Cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T
www.nature.com/scientificreports OPEN Mechanical forces induce an asthma gene signature in healthy airway epithelial cells Ayşe Kılıç1,10, Asher Ameli1,2,10, Jin-Ah Park3,10, Alvin T. Kho4, Kelan Tantisira1, Marc Santolini 1,5, Feixiong Cheng6,7,8, Jennifer A. Mitchel3, Maureen McGill3, Michael J. O’Sullivan3, Margherita De Marzio1,3, Amitabh Sharma1, Scott H. Randell9, Jefrey M. Drazen3, Jefrey J. Fredberg3 & Scott T. Weiss1,3* Bronchospasm compresses the bronchial epithelium, and this compressive stress has been implicated in asthma pathogenesis. However, the molecular mechanisms by which this compressive stress alters pathways relevant to disease are not well understood. Using air-liquid interface cultures of primary human bronchial epithelial cells derived from non-asthmatic donors and asthmatic donors, we applied a compressive stress and then used a network approach to map resulting changes in the molecular interactome. In cells from non-asthmatic donors, compression by itself was sufcient to induce infammatory, late repair, and fbrotic pathways. Remarkably, this molecular profle of non-asthmatic cells after compression recapitulated the profle of asthmatic cells before compression. Together, these results show that even in the absence of any infammatory stimulus, mechanical compression alone is sufcient to induce an asthma-like molecular signature. Bronchial epithelial cells (BECs) form a physical barrier that protects pulmonary airways from inhaled irritants and invading pathogens1,2. Moreover, environmental stimuli such as allergens, pollutants and viruses can induce constriction of the airways3 and thereby expose the bronchial epithelium to compressive mechanical stress. In BECs, this compressive stress induces structural, biophysical, as well as molecular changes4,5, that interact with nearby mesenchyme6 to cause epithelial layer unjamming1, shedding of soluble factors, production of matrix proteins, and activation matrix modifying enzymes, which then act to coordinate infammatory and remodeling processes4,7–10. -

Product Information
Product information NANOG 1-154aa Human, His-tagged, Recombinant, E.coli Cat. No. IBATGP1124 Full name: Homeobox protein NANOG NCBI Accession No.: NP_079141 Synonyms: Homeobox transcription factor Nanog, homeobox transcription factor Nanog-delta 48 Description: NANOG, also known as nanog homeobox, is a member of the homeobox family of DNA binding transcription factors that has been shown to maintain pluripotency of embryonic stem cells. Nanog expression counteracts the differentiation-promoting signals induced by the extrinsic factors LIF, Stat3 and BMP. Once NANOG expression is down-regulated, cell differentiation can proceed. Recombinant human NANOG protein, fused to His- tag at N-terminus, was expressed in E.coli and purified by using conventional chromatography. Form: Liquid. 20mM Tris-HCl buffer (pH8.0) containing 20% glycerol, 1mM DTT Molecular Weight: 19.6 kDa (174aa), confirmed by MALDI-TOF (Molecular weight on SDS-PAGE will appear higher) Purity: > 90% by SDS - PAGE Concentration: 0.25 mg/ml (determined by Bradford assay) 15% SDS-PAGE (3ug) Sequences of amino acids: MGSSHHHHHH SSGLVPRGSH MSVDPACPQS LPCFEASDCK ESSPMPVICG PEENYPSLQM SSAEMPHTET VSPLPSSMDL LIQDSPDSST SPKGKQPTSA EKSVAKKEDK VPVKKQKTRT VFSSTQLCVL NDRFQRQKYL SLQQMQELSN ILNLSYKQVK TWFQNQRMKS KRWQ General references: Clark A T., et al. (2004) Stem Cells. 22:169-179. Chambers I., et al. (2003) Cell Res. 13:499-502. Storage: Can be stored at +4°C short term (1-2 weeks). For long term storage, aliquot and store at -20°C or -70°C. Avoid repeated freezing and thawing cycles. For research use only. This product is not intended or approved for human, diagnostics or veterinary use. Immuno-Biological Laboratories, Inc. (IBL-America) 8201 Central Ave. -

Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation
From THE DEPARTMENT OF MICROBIOLOGY TUMOR AND CELL BIOLOGY (MTC) Karolinska Institutet, Stockholm, Sweden ROLE OF MITOCHONDRIAL RIBOSOMAL PROTEIN S18-2 IN CANCEROGENESIS AND IN REGULATION OF STEMNESS AND DIFFERENTIATION Muhammad Mushtaq Stockholm 2017 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by E-Print AB 2017 © Muhammad Mushtaq, 2017 ISBN 978-91-7676-697-2 Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation THESIS FOR DOCTORAL DEGREE (Ph.D.) By Muhammad Mushtaq Principal Supervisor: Faculty Opponent: Associate Professor Elena Kashuba Professor Pramod Kumar Srivastava Karolinska Institutet University of Connecticut Department of Microbiology Tumor and Cell Center for Immunotherapy of Cancer and Biology (MTC) Infectious Diseases Co-supervisor(s): Examination Board: Professor Sonia Lain Professor Ola Söderberg Karolinska Institutet Uppsala University Department of Microbiology Tumor and Cell Department of Immunology, Genetics and Biology (MTC) Pathology (IGP) Professor George Klein Professor Boris Zhivotovsky Karolinska Institutet Karolinska Institutet Department of Microbiology Tumor and Cell Institute of Environmental Medicine (IMM) Biology (MTC) Professor Lars-Gunnar Larsson Karolinska Institutet Department of Microbiology Tumor and Cell Biology (MTC) Dedicated to my parents ABSTRACT Mitochondria carry their own ribosomes (mitoribosomes) for the translation of mRNA encoded by mitochondrial DNA. The architecture of mitoribosomes is mainly composed of mitochondrial ribosomal proteins (MRPs), which are encoded by nuclear genomic DNA. Emerging experimental evidences reveal that several MRPs are multifunctional and they exhibit important extra-mitochondrial functions, such as involvement in apoptosis, protein biosynthesis and signal transduction. Dysregulations of the MRPs are associated with severe pathological conditions, including cancer. -

Cell Cycle and Pluripotency: Convergence on Octamer‑Binding Transcription Factor 4 (Review)
MOLECULAR MEDICINE REPORTS 16: 6459-6466, 2017 Cell cycle and pluripotency: Convergence on octamer‑binding transcription factor 4 (Review) SHIQI SHE1*, QUCHENG WEI2*, BO KANG1 and YING-JIE WANG1 1State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310003; 2Cardiovascular Key Lab of Zhejiang, Department of Cardiology, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310009, P.R. China Received December 17, 2016; Accepted July 14, 2017 DOI: 10.3892/mmr.2017.7489 Abstract. Embryonic stem cells (ESCs) have unlimited expan- 1. Introduction sion potential and the ability to differentiate into all somatic cell types for regenerative medicine and disease model studies. Embryonic stem cells (ESCs) are characterized by unlimited Octamer-binding transcription factor 4 (OCT4), encoded by proliferation (self-renewal) and the ability to differentiate into the POU domain, class 5, transcription factor 1 gene, is a tran- three primary germ layers, namely the endoderm, mesoderm scription factor vital for maintaining ESC pluripotency and and ectoderm (pluripotency) (1-4). It has been established somatic reprogramming. Many studies have established that that complicated regulatory networks are present in ESCs the cell cycle of ESCs is featured with an abbreviated G1 phase that critically maintain the state of self-renewal and pluri- and a prolonged S phase. Changes in cell cycle dynamics are potency for later development (5,6). Several transcription intimately associated with the state of ESC pluripotency, and factors (TFs), including octamer-binding transcription factor manipulating cell-cycle regulators could enable a controlled 4 (OCT4), SRY-box 2 (SOX2) and homeobox protein NANOG differentiation of ESCs. -

Investigating the Anti-Apoptotic Role of EBV in Endemic Burkitt
Investigating the anti-apoptotic role of EBV in endemic Burkitt lymphoma by LEAH FITZSIMMONS A thesis submitted to The University of Birmingham for the degree of DOCTOR OF PHILOSOPHY School of Cancer Sciences College of Medical and Dental Sciences The University of Birmingham September 2014 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. Abstract Epstein-Barr virus (EBV) has been etiologically associated with Burkitt lymphoma (BL) since its discovery 50 years ago, but despite this long-standing association the precise role of the virus in the pathogenesis of BL remains enigmatic. EBV can be lost spontaneously from EBV-positive BL cell lines, and these EBV-loss clones have been reported to exhibit increased sensitivity to apoptosis. We have confirmed and extended those observations and report that sporadic loss of EBV from BL cells is consistently associated with enhanced sensitivity to apoptosis-inducing agents and conversely, reduced tumorigenicity in vivo. Importantly, reinfection of EBV-loss clones with EBV can restore apoptosis protection, although surprisingly, individual Latency I genes cannot. We also used inducible pro-apoptotic BH3 ligands to investigate Bcl-2-family dependence in BL clones as well as profiling gene expression changes in response to apoptosis induction in EBV- positive versus EBV-loss clones. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Efficient Genome Editing in Multiple Salmonid Cell Lines Using Ribonucleoprotein Complexes
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.03.022038; this version posted April 3, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Efficient genome editing in multiple salmonid cell lines using ribonucleoprotein complexes 2 Remi L. Gratacap1*, Ye Hwa Jin1*, Marina Mantsopoulou1, and Ross D. Houston1** 3 1. The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, 4 Midlothian EH25 9RG, United Kingdom 5 *These authors contributed equally to this work 6 ** Corresponding author: [email protected] 7 ORCID ID: 8 Remi Gratacap: 0000-0001-9853-2205 9 Ye Hwa Jin: 0000-0003-2736-2493 10 Ross Houston: 0000-0003-1805-0762 11 Abstract 12 Infectious and parasitic diseases have major negative economic and animal welfare impacts on 13 aquaculture of salmonid species. Improved knowledge of the functional basis of host response and 14 genetic resistance to these diseases is key to developing preventative and treatment options. Cell 15 lines provide a valuable model to study infectious diseases in salmonids, and genome editing using 16 CRISPR provides an exciting avenue to evaluate the function of specific genes in those systems. 17 While CRISPR/Cas9 has been successfully performed in a Chinook salmon cell line (CHSE-214), 18 there are no reports to date of editing of cell lines derived from the most commercially relevant 19 salmonid species Atlantic salmon and rainbow trout, which are difficult to transduce and therefore 20 edit using lentivirus-mediated methods. -
Type of the Paper (Article
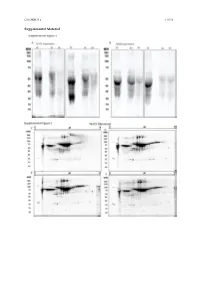
Cells 2020, 9, x 1 of 19 Supplemental Material Cells 2020, 9, x 2 of 19 Figure S1. Secretome enrichment: protocol optimization. 1D SDS-PAGE documentation of washing steps: Culture medium was substituted with FCS-free medium, which was changed every 2 h. The supernatants were then collected, and the proteins isolated and separated in 1D SDS-PAGE ((A) TK173 and (B) TK188). Proteins were stained with Flamingo fluorescent gel stain. Two-dimensional pattern of the proteins isolated from supernatant of TK173, (C) 2 h, (D) 4 h, (E) 6 h, and (F) 8 h after changing to FCS-free medium. (G) Cell secretome collected 24 h after elimination of the contaminating FCS-proteins with different washing steps. Proteins were stained with Flamingo fluorescent gel stain. Cells 2020, 9, x 3 of 19 Figure S2. 2-DE reference maps of secretomes; 150 μg proteins were loaded on an 11 cm IPG strip with a linear pH gradient PI 5–8 for IEF; 12% SDS-polyacrylamide gels were used for the second dimension. Proteins were stained with Flamingo fluorescent gel stain. Identified spots were assigned a number corresponding to that in their table. 2-DE maps from secretome of (A) TK173 control and (B) TGFβ1- treated ones. The 2-DE patterns revealed an alteration of secretome in stimulated TK173. Secretome patterns from TK173 treated with (C) ANG II and (D) PDGF. Cells 2020, 9, x 4 of 19 A Figure S3. Classification of the differentially expressed proteins upon ANG II, TGFβ1, or PDGF treatment in TK173. (A) Bar charts of the cellular component analyzed by STRAP biological function analysis in which the identified proteins from all treatments in both cell types are involved. -
Ep001645626b1*
(19) *EP001645626B1* (11) EP 1 645 626 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 5/06 (2006.01) A61K 48/00 (2006.01) 12.09.2007 Bulletin 2007/37 (21) Application number: 05255932.5 (22) Date of filing: 23.09.2005 (54) Cell line Zelllinie Lignée cellulaire (84) Designated Contracting States: (56) References cited: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR WO-A-01/66781 WO-A-97/10329 HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI US-A- 5 580 777 US-A- 5 770 414 SK TR US-A1- 2003 143 737 (30) Priority: 30.09.2004 GB 0421753 • LITTLEWOOD T D ET AL: "A MODIFIED 23.11.2004 GB 0425767 OESTROGEN RECEPTOR LIGAND-BINDING 20.12.2004 GB 0427830 DOMAIN AS AN IMPROVED SWITCH FOR THE REGULATION OF HETEROLOGOUS PROTEINS" (43) Date of publication of application: NUCLEIC ACIDS RESEARCH, OXFORD 12.04.2006 Bulletin 2006/15 UNIVERSITY PRESS, SURREY, GB, vol. 23, no. 10, 1995, pages 1686-1690, XP002925103 ISSN: (73) Proprietor: Reneuron Limited 0305-1048 Guildford, • GRAY J A ET AL: "PROSPECTS FOR THE Surrey GU2 7AF (GB) CLINICAL APPLICATION OF NEURAL TRANSPLANTATION WITH THE USE OF (72) Inventors: CONDITIONALLY IMMORTALIZED • Sinden, John NEUROEPITHELIAL STEM CELLS" Guildford, PHILOSOPHICAL TRANSACTIONS. ROYAL Surrey GU2 7AF (GB) SOCIETY OF LONDON. BIOLOGICAL SCIENCES, • Pollack, Kenneth ROYAL SOCIETY, LONDON, GB, vol. 354, no. Guildford, 1388, August 1999 (1999-08), pages 1407-1421, Surrey GU2 7AF (GB) XP000865924 ISSN: 0962-8436 • Stroemer, Paul Guildford, Remarks: Surrey GU2 7AF (GB) Thefilecontainstechnicalinformationsubmittedafter the application was filed and not included in this (74) Representative: Jappy, John William Graham specification Gill Jennings & Every LLP Broadgate House 7 Eldon Street London EC2M 7LH (GB) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

Studies Relating to the Differentiation of Human Embryonic Stem Cells
Studies relating to the differentiation of human embryonic stem cells A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Sciences 2015 Georgios Anyfantis List of Figures .................................................................................................... 10 List of Tables ...................................................................................................... 13 Declaration ........................................................................................................ 15 Copyright statement .......................................................................................... 15 Acknowledgements ........................................................................................... 16 Abbreviations .................................................................................................... 17 Chapter 1: Introduction ...................................................................................... 23 1.1 Diabetes Mellitus ......................................................................................... 23 1.2 Pancreatic function and Organogenesis ........................................................ 24 1.2.1 Pancreatic Function .................................................................................... 24 1.2.2 Pancreatic Organogenesis ........................................................................... 26 1.3 Signalling Pathways associated with pancreas development ....................... -

Origin, Characterization and Roles of Matrix Vesicles in Physiological and Pathological Mineralization
Origin, characterization and roles of matrix vesicles in physiological and pathological mineralization. Cyril Thouverey To cite this version: Cyril Thouverey. Origin, characterization and roles of matrix vesicles in physiological and pathological mineralization.. Life Sciences [q-bio]. Université Claude Bernard - Lyon I, 2008. English. tel- 00304214 HAL Id: tel-00304214 https://tel.archives-ouvertes.fr/tel-00304214 Submitted on 22 Jul 2008 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. N° d’ordre: 95-2008 Année 2008 THESE Présentée devant Nencki Institute of Experimental Biology, Polish Academy of Sciences Université Claude Bernard Lyon 1, Pour l’obtention Du DIPLOME DE DOCTORAT (Arrêté du 7 août 2006 et arrêté du 6 janvier 2005) Présentée et soutenue publiquement le 20 juin 2008 Par Cyril Thouverey Origin, characterization and roles of matrix vesicles in physiological and pathological mineralization Directeurs de thèse: Pr René Buchet et Pr Sławomir Pikuła JURY: Mme Françoise Bleicher Mme Joanna Bandorowicz-Pikuła Mme Martime Cohen-Solal Mr René Buchet Mr Sławomir Pikuła Mr Aleksander Sikorski UNIVERSITE CLAUDE BERNARD - LYON I Président de l’Université M. le Professeur L. COLLET Vice-Président du Conseil Scientifique M.