Research 1..9
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Articles Catalytic Cycling in Β-Phosphoglucomutase: a Kinetic
9404 Biochemistry 2005, 44, 9404-9416 Articles Catalytic Cycling in â-Phosphoglucomutase: A Kinetic and Structural Analysis†,‡ Guofeng Zhang, Jianying Dai, Liangbing Wang, and Debra Dunaway-Mariano* Department of Chemistry, UniVersity of New Mexico, Albuquerque, New Mexico 87131-0001 Lee W. Tremblay and Karen N. Allen* Department of Physiology and Biophysics, Boston UniVersity School of Medicine, Boston, Massachusetts 02118-2394 ReceiVed March 26, 2005; ReVised Manuscript ReceiVed May 18, 2005 ABSTRACT: Lactococcus lactis â-phosphoglucomutase (â-PGM) catalyzes the interconversion of â-D-glucose 1-phosphate (â-G1P) and â-D-glucose 6-phosphate (G6P), forming â-D-glucose 1,6-(bis)phosphate (â- G16P) as an intermediate. â-PGM conserves the core domain catalytic scaffold of the phosphatase branch of the HAD (haloalkanoic acid dehalogenase) enzyme superfamily, yet it has evolved to function as a mutase rather than as a phosphatase. This work was carried out to identify the structural basis underlying this diversification of function. In this paper, we examine â-PGM activation by the Mg2+ cofactor, â-PGM activation by Asp8 phosphorylation, and the role of cap domain closure in substrate discrimination. First, the 1.90 Å resolution X-ray crystal structure of the Mg2+-â-PGM complex is examined in the context of + + previously reported structures of the Mg2 -R-D-galactose-1-phosphate-â-PGM, Mg2 -phospho-â-PGM, and Mg2+-â-glucose-6-phosphate-1-phosphorane-â-PGM complexes to identify conformational changes that occur during catalytic turnover. The essential role of Asp8 in nucleophilic catalysis was confirmed by demonstrating that the D8A and D8E mutants are devoid of catalytic activity. -

Diagnosis, Treatment and Follow Up
DOI: 10.1002/jimd.12024 REVIEW International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up Ruqaiah Altassan1,2 | Romain Péanne3,4 | Jaak Jaeken3 | Rita Barone5 | Muad Bidet6 | Delphine Borgel7 | Sandra Brasil8,9 | David Cassiman10 | Anna Cechova11 | David Coman12,13 | Javier Corral14 | Joana Correia15 | María Eugenia de la Morena-Barrio16 | Pascale de Lonlay17 | Vanessa Dos Reis8 | Carlos R Ferreira18,19 | Agata Fiumara5 | Rita Francisco8,9,20 | Hudson Freeze21 | Simone Funke22 | Thatjana Gardeitchik23 | Matthijs Gert4,24 | Muriel Girad25,26 | Marisa Giros27 | Stephanie Grünewald28 | Trinidad Hernández-Caselles29 | Tomas Honzik11 | Marlen Hutter30 | Donna Krasnewich18 | Christina Lam31,32 | Joy Lee33 | Dirk Lefeber23 | Dorinda Marques-da-Silva9,20 | Antonio F Martinez34 | Hossein Moravej35 | Katrin Õunap36,37 | Carlota Pascoal8,9 | Tiffany Pascreau38 | Marc Patterson39,40,41 | Dulce Quelhas14,42 | Kimiyo Raymond43 | Peymaneh Sarkhail44 | Manuel Schiff45 | Małgorzata Seroczynska29 | Mercedes Serrano46 | Nathalie Seta47 | Jolanta Sykut-Cegielska48 | Christian Thiel30 | Federic Tort27 | Mari-Anne Vals49 | Paula Videira20 | Peter Witters50,51 | Renate Zeevaert52 | Eva Morava53,54 1Department of Medical Genetic, Montréal Children's Hospital, Montréal, Québec, Canada 2Department of Medical Genetic, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia 3Department of Human Genetics, KU Leuven, Leuven, Belgium 4LIA GLYCOLAB4CDG (International -

Supplementary Table S1. Table 1. List of Bacterial Strains Used in This Study Suppl
Supplementary Material Supplementary Tables: Supplementary Table S1. Table 1. List of bacterial strains used in this study Supplementary Table S2. List of plasmids used in this study Supplementary Table 3. List of primers used for mutagenesis of P. intermedia Supplementary Table 4. List of primers used for qRT-PCR analysis in P. intermedia Supplementary Table 5. List of the most highly upregulated genes in P. intermedia OxyR mutant Supplementary Table 6. List of the most highly downregulated genes in P. intermedia OxyR mutant Supplementary Table 7. List of the most highly upregulated genes in P. intermedia grown in iron-deplete conditions Supplementary Table 8. List of the most highly downregulated genes in P. intermedia grown in iron-deplete conditions Supplementary Figures: Supplementary Figure 1. Comparison of the genomic loci encoding OxyR in Prevotella species. Supplementary Figure 2. Distribution of SOD and glutathione peroxidase genes within the genus Prevotella. Supplementary Table S1. Bacterial strains Strain Description Source or reference P. intermedia V3147 Wild type OMA14 isolated from the (1) periodontal pocket of a Japanese patient with periodontitis V3203 OMA14 PIOMA14_I_0073(oxyR)::ermF This study E. coli XL-1 Blue Host strain for cloning Stratagene S17-1 RP-4-2-Tc::Mu aph::Tn7 recA, Smr (2) 1 Supplementary Table S2. Plasmids Plasmid Relevant property Source or reference pUC118 Takara pBSSK pNDR-Dual Clonetech pTCB Apr Tcr, E. coli-Bacteroides shuttle vector (3) plasmid pKD954 Contains the Porpyromonas gulae catalase (4) -

Letters to Nature
letters to nature Received 7 July; accepted 21 September 1998. 26. Tronrud, D. E. Conjugate-direction minimization: an improved method for the re®nement of macromolecules. Acta Crystallogr. A 48, 912±916 (1992). 1. Dalbey, R. E., Lively, M. O., Bron, S. & van Dijl, J. M. The chemistry and enzymology of the type 1 27. Wolfe, P. B., Wickner, W. & Goodman, J. M. Sequence of the leader peptidase gene of Escherichia coli signal peptidases. Protein Sci. 6, 1129±1138 (1997). and the orientation of leader peptidase in the bacterial envelope. J. Biol. Chem. 258, 12073±12080 2. Kuo, D. W. et al. Escherichia coli leader peptidase: production of an active form lacking a requirement (1983). for detergent and development of peptide substrates. Arch. Biochem. Biophys. 303, 274±280 (1993). 28. Kraulis, P.G. Molscript: a program to produce both detailed and schematic plots of protein structures. 3. Tschantz, W. R. et al. Characterization of a soluble, catalytically active form of Escherichia coli leader J. Appl. Crystallogr. 24, 946±950 (1991). peptidase: requirement of detergent or phospholipid for optimal activity. Biochemistry 34, 3935±3941 29. Nicholls, A., Sharp, K. A. & Honig, B. Protein folding and association: insights from the interfacial and (1995). the thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Genet. 11, 281±296 (1991). 4. Allsop, A. E. et al.inAnti-Infectives, Recent Advances in Chemistry and Structure-Activity Relationships 30. Meritt, E. A. & Bacon, D. J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277, 505± (eds Bently, P. H. & O'Hanlon, P. J.) 61±72 (R. Soc. Chem., Cambridge, 1997). -

The Reaction Mechanism of Phosphomannomutase in Plants
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector FEBS 18031 FEBS Letters 401 (1997) 35-37 The reaction mechanism of phosphomannomutase in plants Christine Oesterhelt, Claus Schnarrenberger, Wolfgang Gross* Institut für Pflanzenphysiologie und Mikrobiologie, Freie Universität Berlin, Königin-Luise-Str. 12-16a, D-14195 Berlin, Germany Received 11 November 1996 the presence of an excess of GIC-I.6-P2, purified PMM from G. sul- Abstract The enzyme phosphomannomutase catalyzes the phuraria, pig brain, and yeast was incubated with 1 mM GIC-I.6-P2 interconversion of mannose-1-phosphate (Man-l-P) and man- and 0.1 mM Man-l-P for 3 h at room temperature. The reaction nose-6-phosphate (Man-6-P). In mammalian cells the enzyme products were separated by TLC at pH 10 as described [8]. The has to be activated by transfer of a phosphate group from a corresponding regions for Man-l-P, Man-6-P, and Glc-6-P were sugar-1.6-P2 (Guha, S.K. and Rose, Z.B. (1985) Arch. Biochem. scraped off, the sugar phosphates eluted, and identified enzymatically. Biophys. 243, 168). In contrast, in the red alga Galdieria The concentration of Glc-6-P was determined by the addition of Glc- sulphuraria the co-substrate (Man-1.6-P2 or GIC-I.6-P2) is 6-P dehydrogenase and NADP. For Man-6-P determination PGI and PMI were included and for Man-l-P purified PMM from G. sulphu- converted to the corresponding sugar monophosphate while the raria was added. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -
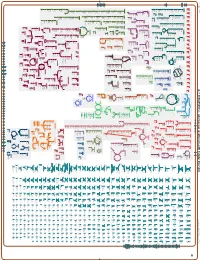
Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000020585Cyc: Alteromonas mediterranea DE Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari pro pro L-fucose GlnK RS01880 FucP RS01650 RS01915 PutP PutP L-fucose pro pro RS01595 Aminoacyl-tRNA Charging Amino Acid Degradation N 6 -(3-methylbut- a nascent peptidoglycan a peptidoglycan UDP-N-acetyl-α-D- a nascent DNA with adenine:7,8- (4R)-4-hydroxy- a peptidoglycan a [protein]-L- queuosine at an [amino-group a sulfurated + a nucleoside an L-asparaginyl- tRNA charging L-valine degradation I 2-en-1-yl)- γ γ muramoyl-L-alanyl- NADPH NAD a [glutamine- dihydro-8- dimer (meso- glutamate-O 5 Fermentation mixed acid fermentation L-isoleucine degradation I Alcohol Degradation with (L-alanyl- -D- with (L-alanyl- -D- peptidoglycan ATP 2-oxoglutarate RNA glu position 34 Asn carrier protein]- L-leucine degradation I L-threonine degradation I L-arginine degradation [sulfur carrier] 37 synthetase]- triphosphate [tRNA ] adenosine glutamyl-meso-2,6- glutamyl-meso-2,6- γ-D-glutamyl-meso- -
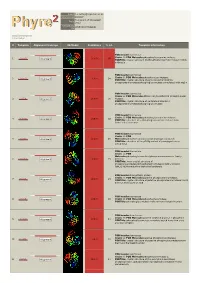
Phyre 2 Results for P0C0V7
Email [email protected] Description P0C0V7 Tue Jul 17 17:05:08 BST Date 2012 Unique Job c896433374b5eb81 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:isomerase Chain: B: PDB Molecule:phosphoglucosamine mutase; 1 c3pdkB_ 100.0 68 Alignment PDBTitle: crystal structure of phosphoglucosamine mutase from b. anthracis PDB header:isomerase Chain: B: PDB Molecule:phospho-sugar mutase; 2 c1wqaB_ 100.0 34 Alignment PDBTitle: crystal structure of pyrococcus horikoshii2 phosphomannomutase/phosphoglucomutase complexed with mg2+ PDB header:isomerase Chain: A: PDB Molecule:455aa long hypothetical phospho-sugar 3 c2f7lA_ Alignment 100.0 30 mutase; PDBTitle: crystal structure of sulfolobus tokodaii2 phosphomannomutase/phosphoglucomutase PDB header:isomerase Chain: B: PDB Molecule:phosphoglucosamine mutase; 4 c3i3wB_ 100.0 38 Alignment PDBTitle: structure of a phosphoglucosamine mutase from francisella tularensis PDB header:isomerase Chain: A: PDB 5 c3c04A_ Alignment 100.0 26 Molecule:phosphomannomutase/phosphoglucomutase; PDBTitle: structure of the p368g mutant of pmm/pgm from p. aeruginosa PDB header:isomerase Chain: A: PDB Molecule:phosphoglucomutase/phosphomannomutase family 6 c3uw2A_ Alignment 100.0 25 protein; PDBTitle: x-ray crystal structure of phosphoglucomutase/phosphomannomutase2 family protein (bth_i1489)from burkholderia thailandensis PDB header:biosynthetic protein Chain: A: PDB Molecule:putative phosphomannomutase; 7 c1tuoA_ 100.0 25 Alignment PDBTitle: -

12) United States Patent (10
US007635572B2 (12) UnitedO States Patent (10) Patent No.: US 7,635,572 B2 Zhou et al. (45) Date of Patent: Dec. 22, 2009 (54) METHODS FOR CONDUCTING ASSAYS FOR 5,506,121 A 4/1996 Skerra et al. ENZYME ACTIVITY ON PROTEIN 5,510,270 A 4/1996 Fodor et al. MICROARRAYS 5,512,492 A 4/1996 Herron et al. 5,516,635 A 5/1996 Ekins et al. (75) Inventors: Fang X. Zhou, New Haven, CT (US); 5,532,128 A 7/1996 Eggers Barry Schweitzer, Cheshire, CT (US) 5,538,897 A 7/1996 Yates, III et al. s s 5,541,070 A 7/1996 Kauvar (73) Assignee: Life Technologies Corporation, .. S.E. al Carlsbad, CA (US) 5,585,069 A 12/1996 Zanzucchi et al. 5,585,639 A 12/1996 Dorsel et al. (*) Notice: Subject to any disclaimer, the term of this 5,593,838 A 1/1997 Zanzucchi et al. patent is extended or adjusted under 35 5,605,662 A 2f1997 Heller et al. U.S.C. 154(b) by 0 days. 5,620,850 A 4/1997 Bamdad et al. 5,624,711 A 4/1997 Sundberg et al. (21) Appl. No.: 10/865,431 5,627,369 A 5/1997 Vestal et al. 5,629,213 A 5/1997 Kornguth et al. (22) Filed: Jun. 9, 2004 (Continued) (65) Prior Publication Data FOREIGN PATENT DOCUMENTS US 2005/O118665 A1 Jun. 2, 2005 EP 596421 10, 1993 EP 0619321 12/1994 (51) Int. Cl. EP O664452 7, 1995 CI2O 1/50 (2006.01) EP O818467 1, 1998 (52) U.S. -

Supplementary Materials
Supplementary Materials OH 8 H H8 H C N OH 3 7 3 2 4 H1 1 5 O H4 O 6 H6’ H3 OH H6 H1’ H2 H5 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 ppm 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 ppm Figure S1. 1D 1H spectrum of 1-dglcnac and signal assignments ppm 2.0 H8 2.5 OH 8 H H C N OH 3 7 3 2 4 3.0 1 5 H1 O H5 O 6 H4 H3 OH 3.5 H6 H2 H6’ H1’ 4.0 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 2.0 ppm Figure S2. 1H-1H TOCSY 2D NMR spectra for 1-dglcnac and signal assignments ppm 3.0 H1 3.2 H5 H4 3.4 H3 3.6 H6 H6’ H2 3.8 H1’ 4.0 4.1 4.0 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 3.1 3.0 ppm Figure S3. 1H-1H COSY 2D NMR spectra for 1-dglcnac and signal assignment ppm -0.02 -0.01 0.00 0.01 0.02 H1’ H6’ H2 H6 H3 H4 H5 H1 3.9 3.8 3.7 3.6 3.5 3.4 3.3 3.2 3.1 ppm Figure S4. 1H-1H JRES 2D NMR spectra for 1-dglcnac and signal assignments ppm 20 C8(22.9) 40 OH 8 H H C N OH C2(53.2) 3 7 3 2 4 60 1 5 C6(63.2) C6(63.2) O 6 C1(69.2) C1(69.2) O C4(72.6) C3(77.0) OH 80 C5(82.7) 100 4.0 3.8 3.6 3.4 3.2 3.0 2.8 2.6 2.4 2.2 2.0 ppm Figure S5. -

REVIEW ARTICLE Congenital Disorders of Glycosylation
0031-3998/02/5205-0618 PEDIATRIC RESEARCH Vol. 52, No. 5, 2002 Copyright © 2002 International Pediatric Research Foundation, Inc. Printed in U.S.A. REVIEW ARTICLE Congenital Disorders of Glycosylation: A Review STEPHANIE GRÜNEWALD, GERT MATTHIJS, AND JAAK JAEKEN Children’s University Hospital Essen, 45122 Essen, Germany [S.G.], and Centers for Human Genetics [G.M.] and Metabolic Disease [J.J.], Katholieke Universiteit Leuven, 3000 Leuven, Belgium ABSTRACT Congenital disorders of glycosylation (CDGs) are a rapidly Abbreviations growing group of inherited disorders caused by defects in the CDG, congenital disorder(s) of glycosylation, formerly synthesis and processing of the asparagine(ASN)-linked oligo- carbohydrate-deficient glycoprotein syndrome(s) saccharides of glycoproteins. The first CDG patients were de- PMM, phosphomannomutase scribed in 1980. Fifteen years later, a phosphomannomutase MPI, phosphomannose isomerase deficiency was found as the basis of the most frequent type, ER, endoplasmic reticulum CDG-Ia. In recent years several novel types have been identified. IEF, isoelectrofocusing The N-glycosylation pathway is highly conserved from yeast to LLO, lipid-linked oligosaccharide human, and the rapid progress in this field can largely be Glc, glucose attributed to the systematic application of the knowledge of yeast Man, mannose mutants. Up to now, eight diseases have been characterized, GlcNAc, N-acetylglucosamine resulting from enzyme or transport defects in the cytosol, endo- ALG 6, ␣-1,3-glucosyltransferase plasmic reticulum, or Golgi compartment. CDGs affect all organs ALG 3, ␣-1,3-mannosyltransferase and particularly the CNS, except for CDG-Ib, which is mainly a ASN, asparagine hepatic-intestinal disease. (Pediatr Res 52: 618–624, 2002) GDP, guanosine diphosphate The biosynthesis of asparagine (ASN)-linked oligosaccha- resistance, host defense, and antigenicity.