NHC-Supported Mixed Halohydrides of Aluminium and Related Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 3.3 Special Provisions Applicable to Certain Articles Or Substances
Chapter 3.3 Special provisions applicable to certain articles or substances 3.3.1 When Column (6) of Table A of Chapter 3.2 indicates that a special provision is relevant to a substance or article, the meaning and requirements of that special provision are as set forth below. 16 Samples of new or existing explosive substances or articles may be carried as directed by the competent authorities (see 2.2.1.1.3) for purposes including: testing, classification, research and development, qual- ity control, or as a commercial sample. Explosive samples which are not wetted or desensitized shall be limited to 10 kg in small packages as specified by the competent authorities. Explosive samples which are wetted or desensitized shall be limited to 25 kg. 23 Even though this substance has a flammability hazard, it only exhibits such hazard under extreme fire conditions in confined areas. 32 This substance is not subject to the requirements of RID when in any other form. 37 This substance is not subject to the requirements of RID when coated. 38 This substance is not subject to the requirements of RID when it contains not more than 0.1% calcium carbide. 39 This substance is not subject to the requirements of RID when it contains less than 30% or not less than 90% silicon. 43 When offered for carriage as pesticides, these substances shall be carried under the relevant pesticide entry and in accordance with the relevant pesticide provisions (see 2.2.61.1.10 to 2.2.61.1.11.2). 45 Antimony sulphides and oxides which contain not more than 0.5% of arsenic calculated on the total mass are not subject to the requirements of RID. -

NBO Applications, 2020
NBO Bibliography 2020 2531 publications – Revised and compiled by Ariel Andrea on Aug. 9, 2021 Aarabi, M.; Gholami, S.; Grabowski, S. J. S-H ... O and O-H ... O Hydrogen Bonds-Comparison of Dimers of Thiocarboxylic and Carboxylic Acids Chemphyschem, (21): 1653-1664 2020. 10.1002/cphc.202000131 Aarthi, K. V.; Rajagopal, H.; Muthu, S.; Jayanthi, V.; Girija, R. Quantum chemical calculations, spectroscopic investigation and molecular docking analysis of 4-chloro- N-methylpyridine-2-carboxamide Journal of Molecular Structure, (1210) 2020. 10.1016/j.molstruc.2020.128053 Abad, N.; Lgaz, H.; Atioglu, Z.; Akkurt, M.; Mague, J. T.; Ali, I. H.; Chung, I. M.; Salghi, R.; Essassi, E.; Ramli, Y. Synthesis, crystal structure, hirshfeld surface analysis, DFT computations and molecular dynamics study of 2-(benzyloxy)-3-phenylquinoxaline Journal of Molecular Structure, (1221) 2020. 10.1016/j.molstruc.2020.128727 Abbenseth, J.; Wtjen, F.; Finger, M.; Schneider, S. The Metaphosphite (PO2-) Anion as a Ligand Angewandte Chemie-International Edition, (59): 23574-23578 2020. 10.1002/anie.202011750 Abbenseth, J.; Goicoechea, J. M. Recent developments in the chemistry of non-trigonal pnictogen pincer compounds: from bonding to catalysis Chemical Science, (11): 9728-9740 2020. 10.1039/d0sc03819a Abbenseth, J.; Schneider, S. A Terminal Chlorophosphinidene Complex Zeitschrift Fur Anorganische Und Allgemeine Chemie, (646): 565-569 2020. 10.1002/zaac.202000010 Abbiche, K.; Acharjee, N.; Salah, M.; Hilali, M.; Laknifli, A.; Komiha, N.; Marakchi, K. Unveiling the mechanism and selectivity of 3+2 cycloaddition reactions of benzonitrile oxide to ethyl trans-cinnamate, ethyl crotonate and trans-2-penten-1-ol through DFT analysis Journal of Molecular Modeling, (26) 2020. -

An Investigation of the Crystal Growth of Heavy Sulfides in Supercritical
AN ABSTRACT OF THE THESIS OF LEROY CRAWFORD LEWIS for the Ph. D. (Name) (Degree) in CHEMISTRY presented on (Major) (Date) Title: AN INVESTIGATION OF THE CRYSTAL GROWTH OF HEAVY SULFIDES IN SUPERCRITICAL HYDROGEN SULFIDE Abstract approved Redacted for privacy Dr. WilliarriIJ. Fredericks Solubility studies on the heavy metal sulfides in liquid hydrogen sulfide at room temperature were carried out using the isopiestic method. The results were compared with earlier work and with a theoretical result based on Raoult's Law. A relative order for the solubilities of sulfur and the sulfides of tin, lead, mercury, iron, zinc, antimony, arsenic, silver, and cadmium was determined and found to agree with the theoretical result. Hydrogen sulfide is a strong enough oxidizing agent to oxidize stannous sulfide to stannic sulfide in neutral or basic solution (with triethylamine added). In basic solution antimony trisulfide is oxi- dized to antimony pentasulfide. In basic solution cadmium sulfide apparently forms a bisulfide complex in which three moles of bisul- fide ion are bonded to one mole of cadmium sulfide. Measurements were made extending the range over which the volumetric properties of hydrogen sulfide have been investigated to 220 °C and 2000 atm. A virial expression in density was used to represent the data. Good agreement, over the entire range investi- gated, between the virial expressions, earlier work, and the theorem of corresponding states was found. Electrical measurements were made on supercritical hydro- gen sulfide over the density range of 10 -24 moles per liter and at temperatures from the critical temperature to 220 °C. Dielectric constant measurements were represented by a dielectric virial ex- pression. -
![Triazene (H2NNNH) Or Triimide (HNHNNH) Markofçrstel,[A, D] Yetsedaw A](https://docslib.b-cdn.net/cover/4724/triazene-h2nnnh-or-triimide-hnhnnh-markof%C3%A7rstel-a-d-yetsedaw-a-184724.webp)
Triazene (H2NNNH) Or Triimide (HNHNNH) Markofçrstel,[A, D] Yetsedaw A
DOI:10.1002/cphc.201600414 Articles On the Formation of N3H3 Isomers in Irradiated Ammonia Bearing Ices:Triazene (H2NNNH) or Triimide (HNHNNH) MarkoFçrstel,[a, d] Yetsedaw A. Tsegaw,[b] Pavlo Maksyutenko,[a, d] Alexander M. Mebel,[c] Wolfram Sander,[b] and Ralf I. Kaiser*[a, d] The remarkable versatility of triazenesinsynthesis, polymer theoretical studies with our novel detection scheme of photo- chemistry and pharmacology has led to numerousexperimen- ionization-driven reflectron time-of-flight mass spectroscopy tal and theoretical studies.Surprisingly,only very little is we can obtain information on the isomersoftriazene formed known aboutthe most fundamental triazene:the parentmole- in the films. Using isotopically labeled starting material, we can cule with the chemical formula N3H3.Here we observe molecu- additionally gain insightinthe formation pathways of the iso- lar,isolated N3H3 in the gas phase after it sublimes from ener- mers of N3H3 under investigation and identify the isomers getically processed ammonia and nitrogen films. Combining formedastriazene (H2NNNH) andpossibly triimide(HNHNNH). 1. Introduction During the last decades, triazenes—a class of organic mole- life time of at least 1mswas also inferred as an intermediate cules carrying the =N N=N moiety—have received substan- in the radiolysis of an aqueous solution of hydrazine based on À À tial attention both from the theoretical and organic chemistry asingle absorption feature at 230 nm.[6] The cyclic isomer of [1] communities. Derived from cis-and trans-triazene (HN=NNH2 ; triazene, cyclotriazane, was first reported crystallographically in Scheme1), the substituted counterparts have significant appli- zeolite A, where it was stabilized by asilver cation as [1a,c] [1d] + [7] + cations in synthetic chemistry, polymer science, and phar- Ag(N3H3) . -
Chapter 13.Pptx
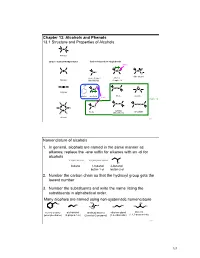
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Studies on New Vasodilators, Ws-1228 a and B Ii. Structure and Synthesis
VOL. XXXV NO. 2 THE JOURNAL OF ANTIBIOTICS 157 STUDIES ON NEW VASODILATORS, WS-1228 A AND B II. STRUCTURE AND SYNTHESIS HIROKAZU TANAKA, KEIZO YOSHIDA, YOSHIKUNI ITOH and HIROSHI IMANAKA Fermentation Research Laboratories, Fujisawa Pharmaceutical Co., Ltd., Osaka, Japan (Received for publication October 26, 1981) The structure of new hypotensive vasodilators, WS-1228 A and B, produced by Strepto- myces aureofaciens, were determined as I and 2, respectively, on the basis of their spectral and chemical evidences. WS-1228 A (1), having N-hydroxytriazene moiety, was synthesized from (E,E,E)-2,4,7- undecatrienal (4) by condensation with hydrazine hydrate followed by nitrosation. In the course of screening for new biologically active compounds, we found that Streptomyces aureofaciens produces hypotensive vasodilators designated as WS-1228 A (1) and B (2). Taxonomy, isolation and characterization of these compounds have been reported in the preceding papery. This report describes the structure elucidations of WS-1228 A (1) and B (2) and the synthesis of 1. WS-1228 A (1) WS-1228 B (2) WS-1228 A (1) was isolated as yellow needles [mp 100 - 102°C (dec.)] which showed a positive color reaction to ferric chloride reagent. Elemental analysis and mass spectrum established the molecular formula of 1 as C11H17N3O. Absorption bands at 1612 and 1580 cm-1 in its IR spectrum (Fig. 1) and at 300 nm in its UV spectrum suggested the presence of the triene oxime moiety. The PMR spectrum (Fig. Fig. 1. IR spectrum of WS-1228 A (1) (Nujol). 158 THE JOURNAL OF ANTIBIOTICS FEB. -

United States Patent Office 2,807,613
United States Patent Office 2,807,613 Patented Sept. 24, 1957 s s 2 2,807,613 nol, where it may exist in the form of a hemiformal, or PREPARATION OF 6-METHYL-6-PHENYLTETRA of a revertible polymer. Usually formaldehyde is used in HYDRO-1,3-OXAZINES excess based on molar proportions referred to the a Claude 5. Schmide, Moorestown, and Richard C. Maas 5 methylstyrene, proportions from about 1.5:1 to 5:1 being field, Haddon afield, N. J., assignors to Rohm & Hiaas practical. Of course, with less than a 2:1 proportion Company, Philadelphia, Pa., a corporatica of Delaware unreacted starting materials may be present in the react ing mixture, Preferred proportions are from 2:1 to 4:1. No Drawing. Application April 3, 1955, Ammonia may be supplied as a gas or as an aqueous Serial No. 577,944 solution or in the form of ammonium chloride or bromide. 7 Claims. (C. 260-244) 0 Of course, if ammonia or ammonium hydroxide is used, This invention deals with a method for improving yields it will react with the hydrochloric or hydrobromic acid of 6-methyl-6-phenyltetrahydro-1,3-oxazines when made which is added as catalyst. The same final result is ob from an O-methylstyrene, formaldehyde, and ammonia. tained by use of the preformed ammonium halide, which In United States Patent 2,647,117, there is described 5 Supplies both the ammonia and the catalyst. The amount the reaction of olefins, including c-methylstyrene, with of ammonia or ammonium compound is usually at least ammonia and formaldehyde in the presence of hydrogen equivalent to the c-methylstyrene and may be in consid -chloride as a catalyst. -

Isomorphous Substitution of Aluminium for Silicon in Tobermoritic Structure
Isomorphous Substitution of Aluminium for Silicon in Tobermoritic Structure. I. The Mixtures of Different Forms of Silicon Dioxide and of Different Compounds of Aluminium J. PETROVIÖ, V. RUSNÁK and L. ŠTEVULA Institute of Inorganic Chemistry, Slovak Academy of Sciences, Bratislava 9 Received July 2, 1968 The isomorphous substitution of Al(III) for Si(IV) in tobermoritic structure was examined by X-ray phase analysis and DTA. It has been found that the extent of the substitution depends on the materials used. The samples were prepared using different forms of silica-either ^-quartz, quartz-glass, Si02-gel or aerosil. Gibbsite, boehmite, dehydrated kaolinite, kaolinite, corundum and 7-AI2O3 were used as sources of aluminium. The samples were heated in an autoclave at 150, 180 and 200°C for 10, 24 and in some cases for 48 hours. Tobermorite can be synthesized from calcium oxide and silica under hydrothermal conditions. Up to temperatures of about 110°C it is stable, at higher temperatures and under hydrothermal conditions it is formed as an unstable compound. When the reaction is allowed to proceed for a longer time, or when it takes place at higher tem perature, another stable calcium hydrosilicate arises. At lower temperature a calcium hydrosilicate with tobermoritic structure is formed. It has been found that tobermori te is produced on autoclaving building materials containing lime and some materials with high SÍO2 content. It is also formed from anhydrous dicalcium silicates in the course of setting of cement. Under different conditions, compounds with tobermoritic structure arise which, however, differ in the amount or lime and water in the molecu le, as well as in the arrangement of the structure — e.g. -

Absence of Skin Sensitivity to Oxides of Aluminium, Silicon, Titanium Or Zirconium in Patients With
Gut1996;39:231-233 231 Absence of skin sensitivity to oxides of aluminium, Silicon, titanium or zirconium in patients with Crohn's disease Gut: first published as 10.1136/gut.39.2.231 on 1 August 1996. Downloaded from J C W Lee, S Halpem, D G Lowe, A Forbes, J E Lennard-Jones Abstract obstructive lymphadenopathy. It has been Background-Some metallic compounds, proposed that this is caused by fibrosis of the especially of zirconium, can cause cell afferent lymphatics as a result of absorption of mediated granulomatous inflammation of microparticles of silica and alumino-silicates the skin. Pigment granules containing through the skin where people walk barefoot compounds of aluminium, silicon, and on certain types of soil. Particles containing titanium have been observed within silica, titanium, and aluminium are present in macrophages in the wall of the small microgranulomata within inguinal lymph intestine in health and in Crohn's disease. nodes of sufferers.6 Granulomata also develop Zirconium compounds can be ingested in in response to intradermal injection ofcolloidal toothpaste. silica in healthy subjects but these are foreign Aim-To determine in a pilot study if body granulomata and are clearly distinguish- granulomatous sensitivity can be detected able from the cell mediated response to small to compounds of these metals or silicon quantities of zirconium lactate.7 after injection into the skin of patients As metals and minerals are ubiquitous in the with Crohn's disease. community, a hypersensitivity to these sub- Subjects-Eight patients with Crohn's stances in some people rather than a direct disease known to have had granulomata in toxic effect is the most probable pathogenetic the intestine and not currently treated mechanism by which they may contribute to with corticosteroids, and two healthy disease. -

Download The
COORDINATION COMPOUNDS OF ALKYL GALLIUM HYDRIDES by VICTOR GRAHAM WIEBE B.Sc. (Hons.) University of British Columbia 1966 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE In The Department of Chemistry We accept this thesis as conforming to the required standard The University of British Columbia June 1968 In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and Study. I further agree that permission for extensive copying of this thesis for scholarly purposes may be granted by the Head of my Department or by hits representatives. It is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. Department of The University of British Columbia Vancouver 8, Canada - ii - Abstract Although the organo hydride derivatives of boron and aluminum are well characterized^little work has been reported on the corresponding gallium systems. The present study was initiated to determine the relative stabilities and reactivity of organo gallium hydride derivatives as compared with the stabilities and reactions of the corresponding compounds of boron and aluminum. Various preparative routes to this new class of gallium compounds have been investigated. These include the use of organo-mercury, organo-lithium and lithium hydride derivatives in reactions with gallium hydride and gallium alkyl compounds and their halogen substituted derivatives: Me3NGaH3 + HgR2 >- Me3NGaH2R + l/2Hg + 1/2H2 Me3NGaH2Cl + LiR y Me3NGaH2R + LiCl Me3NGaR2Cl + LiH • Me3NGaHR2 + LiCl A fourth preparative method involves disproportionation reactions between gallium hydride compounds and organo gallium compounds to yield the mixed organo hydride derivatives. -

Supporting Information For: Solution Phase Synthesis of Indium Gallium Phosphide Alloy Nanowires Nikolay Kornienko , Desiré D
Supporting Information for: Solution Phase Synthesis of Indium Gallium Phosphide Alloy Nanowires Nikolay Kornienko1, Desiré D. Whitmore1, Yi Yu1, Stephen R. Leone1,2,4, Peidong Yang*1,3,5,6 1Department of Chemistry, 2Department of Physics and 3Department of Materials Science Engineering, University of California, Berkeley 94720, United States 4Chemical Sciences Division and 5Materials Sciences Division, Lawrence Berkeley National Lab, Berkeley CA 94720, United States 6Kavli Energy Nanosciences Institute, Berkeley, California 94720, United States * Correspondence to: [email protected] Table of Contents: Figure S1. TEM In/Ga growth seeds ............................................................................................................................. 2 Figure S2. TEM of InP and GaP NWs ............................................................................................................................. 2 Figure S3. Composition and diameter control ......................................................................................................... 3 Figure S4. XRD of InxGa1‐xP NWs .................................................................................................................................. 4 Figure S5. Electron diffraction of InxGa1‐xP NWs ................................................................................................... 5 Figure S6. Raman spectra of InP and GaP .................................................................................................................. 6