Next-Generation Sequencing Identi Es Seasonal And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Genera of Bambusoideae (Gramineae) in the Southeastern United States Gordon C
Eastern Illinois University The Keep Faculty Research & Creative Activity Biological Sciences January 1988 The genera of Bambusoideae (Gramineae) in the southeastern United States Gordon C. Tucker Eastern Illinois University, [email protected] Follow this and additional works at: http://thekeep.eiu.edu/bio_fac Part of the Biology Commons Recommended Citation Tucker, Gordon C., "The eg nera of Bambusoideae (Gramineae) in the southeastern United States" (1988). Faculty Research & Creative Activity. 181. http://thekeep.eiu.edu/bio_fac/181 This Article is brought to you for free and open access by the Biological Sciences at The Keep. It has been accepted for inclusion in Faculty Research & Creative Activity by an authorized administrator of The Keep. For more information, please contact [email protected]. TUCKER, BAMBUSOIDEAE 239 THE GENERA OF BAMBUSOIDEAE (GRAMINEAE) IN THE SOUTHEASTERN UNITED STATESu GoRDON C. T ucKER3 Subfamily BAMBUSOIDEAE Ascherson & Graebner, Synop. Mitteleurop. Fl. 2: 769. 1902. Perennial or annual herbs or woody plants of tropical or temperate forests and wetlands. Rhizomes present or lacking. Stems erect or decumbent (some times rooting at the lower nodes); nodes glabrous, pubescent, or puberulent. Leaves several to many, glabrous to sparsely pubescent (microhairs bicellular); leaf sheaths about as long as the blades, open for over tf2 their length, glabrous; ligules wider than long, entire or fimbriate; blades petiolate or sessile, elliptic to linear, acute to acuminate, the primary veins parallel to-or forming an angle of 5-10• wi th-the midvein, transverse veinlets numerous, usually con spicuous, giving leaf surface a tessellate appearance; chlorenchyma not radiate (i.e., non-kranz; photosynthetic pathway C.,). -

Morphological, Anatomical, and Taxonomic Studies in Anomochloa and Streptochaeta (Poaceae: Bambusoideae)
SMITHSONIAN CONTRIBUTIONS TO BOTANY NUMBER 68 Morphological, Anatomical, and Taxonomic Studies in Anomochloa and Streptochaeta (Poaceae: Bambusoideae) Emmet J. Judziewicz and Thomas R. Soderstrom SMITHSONIAN INSTITUTION PRESS Washington, D.C. 1989 ABSTRACT Judziewicz, Emmet J., and Thomas R. Soderstrom. Morphological, Anatomical, and Taxonomic Studies in Anomochloa and Streptochaeta (Poaceae: Bambusoideae). Smithsonian Contributions to Botany, number 68,52 pages, 24 figures, 1 table, 1989.-Although resembling the core group of the bambusoid grasses in many features of leaf anatomy, the Neotropical rainforest grass genera Anomochloa and Streptochaeta share characters that are unusual in the subfamily: lack of ligules, exceptionally long microhairs with an unusual morphology, a distinctive leaf blade midrib structure, and 5-nerved coleoptiles. Both genera also possess inflorescences that are difficult to interpret in conventional agrostological terms. Anomochloa is monotypic, and A. marantoidea, described in 1851 by Adolphe Brongniart from cultivated material of uncertain provenance, was rediscovered in 1976 in the wet forests of coastal Bahia, Brazil. The inflorescence terminates in a spikelet and bears along its rachis several scorpioid cyme-like partial inflorescences. Each axis of a partial inflorescence is subtended by a keeled bract and bears as its first appendages two tiny, unvascularized bracteoles attached at slightly different levels. The spikelets are composed of an axis that bears two bracts and terminates in a flower. The lower, chlorophyllous, deciduous spikelet bract is separated from the coriaceous, persistent, corniculate upper bract by a cylindrical, indurate internode. The flower consists of a low membrane surmounted by a dense ring of brown cilia (perigonate annulus) surrounding the andrecium of four stamens, and an ovary bearing a single hispid stigma. -

ABSTRACTS 117 Systematics Section, BSA / ASPT / IOPB
Systematics Section, BSA / ASPT / IOPB 466 HARDY, CHRISTOPHER R.1,2*, JERROLD I DAVIS1, breeding system. This effectively reproductively isolates the species. ROBERT B. FADEN3, AND DENNIS W. STEVENSON1,2 Previous studies have provided extensive genetic, phylogenetic and 1Bailey Hortorium, Cornell University, Ithaca, NY 14853; 2New York natural selection data which allow for a rare opportunity to now Botanical Garden, Bronx, NY 10458; 3Dept. of Botany, National study and interpret ontogenetic changes as sources of evolutionary Museum of Natural History, Smithsonian Institution, Washington, novelties in floral form. Three populations of M. cardinalis and four DC 20560 populations of M. lewisii (representing both described races) were studied from initiation of floral apex to anthesis using SEM and light Phylogenetics of Cochliostema, Geogenanthus, and microscopy. Allometric analyses were conducted on data derived an undescribed genus (Commelinaceae) using from floral organs. Sympatric populations of the species from morphology and DNA sequence data from 26S, 5S- Yosemite National Park were compared. Calyces of M. lewisii initi- NTS, rbcL, and trnL-F loci ate later than those of M. cardinalis relative to the inner whorls, and sepals are taller and more acute. Relative times of initiation of phylogenetic study was conducted on a group of three small petals, sepals and pistil are similar in both species. Petal shapes dif- genera of neotropical Commelinaceae that exhibit a variety fer between species throughout development. Corolla aperture of unusual floral morphologies and habits. Morphological A shape becomes dorso-ventrally narrow during development of M. characters and DNA sequence data from plastid (rbcL, trnL-F) and lewisii, and laterally narrow in M. -

Literaturverzeichnis
Literaturverzeichnis Abaimov, A.P., 2010: Geographical Distribution and Ackerly, D.D., 2009: Evolution, origin and age of Genetics of Siberian Larch Species. In Osawa, A., line ages in the Californian and Mediterranean flo- Zyryanova, O.A., Matsuura, Y., Kajimoto, T. & ras. Journal of Biogeography 36, 1221–1233. Wein, R.W. (eds.), Permafrost Ecosystems. Sibe- Acocks, J.P.H., 1988: Veld Types of South Africa. 3rd rian Larch Forests. Ecological Studies 209, 41–58. Edition. Botanical Research Institute, Pretoria, Abbadie, L., Gignoux, J., Le Roux, X. & Lepage, M. 146 pp. (eds.), 2006: Lamto. Structure, Functioning, and Adam, P., 1990: Saltmarsh Ecology. Cambridge Uni- Dynamics of a Savanna Ecosystem. Ecological Stu- versity Press. Cambridge, 461 pp. dies 179, 415 pp. Adam, P., 1994: Australian Rainforests. Oxford Bio- Abbott, R.J. & Brochmann, C., 2003: History and geography Series No. 6 (Oxford University Press), evolution of the arctic flora: in the footsteps of Eric 308 pp. Hultén. Molecular Ecology 12, 299–313. Adam, P., 1994: Saltmarsh and mangrove. In Groves, Abbott, R.J. & Comes, H.P., 2004: Evolution in the R.H. (ed.), Australian Vegetation. 2nd Edition. Arctic: a phylogeographic analysis of the circu- Cambridge University Press, Melbourne, pp. marctic plant Saxifraga oppositifolia (Purple Saxi- 395–435. frage). New Phytologist 161, 211–224. Adame, M.F., Neil, D., Wright, S.F. & Lovelock, C.E., Abbott, R.J., Chapman, H.M., Crawford, R.M.M. & 2010: Sedimentation within and among mangrove Forbes, D.G., 1995: Molecular diversity and deri- forests along a gradient of geomorphological set- vations of populations of Silene acaulis and Saxi- tings. -

Cage on the Sea
Cage on the Sea Kaoru Ohno Translated by Giles Murray http://bentobooks.com CAGE ON THE SEA Originally published as Zekkai Misshitsu Copyright © 1998 by Kaoru Ohno Shinchosha Publishing Co., Ltd., Tokyo English translation © 2014 by Giles Murray Edited by Joseph Reeder Cover design and maps by Kasia Bytnerowicz All rights reserved. No portion of this book in excess of fair use considerations may be reproduced or transmitted in any form or by any means without written permission from the copyright holders. Published 2014 by Bento Books, Inc. Austin, Texas 78732 bentobooks.com ISBN 978-0-9839513-8-4 Printed in the United States of America First edition, March 2014 Cage on the Sea Nemuro Kesennuma Kanuma Fukushima Tokyo Choshi Mt. Fuji Yokohama Misaki Shizuoka Oshima Hachijo Jima Urracas Chichi Jima Iwo Jima Agrihan Pagan Sarigan Mariana Anatahan Islands Saipan Caroline Islands Guam American Canadian Tick Point Bath Brook Kaihomaru Hut First Branch Pine Tree Oce Brook Hyosukemaru Hut Anatahan Ginza B-29 Second Branch Wreckage Oce Company Garden Hospital Hill Sonson Village Sulphur Pits Raft Beach Wall of Rock Misery Brook Breadfruit Brook Fool’s Entrance Brother Beach Velvetleaf Factory Eight Palms Bonito Factory Chapter Two Washed Ashore May 24, 1944 Midnight hrongs of hand-pulled carts hurried back and forth through Tthe darkness, ferrying supplies to the ships from the warehouse beside the wharf. The beacon lights marking the entrance to Yo- kohama harbor and the lights of the customs office had all been doused. Only the faintest wisps of light escaped from the open doors of the warehouse and the holds of the docked ships. -

Majestätisches Japan Motorrad Tour
Majestätisches Japan Land der aufgehenden Sonne 26. September — 11. Oktober, 2020 27. September — 12. Oktober, 2021 | 16. Oktober - 31. Oktober, 2021 Japan ist für viele Touristen eines der begehrtesten Reiseziele. Bei IMTBIKE haben wir eine Motorradroute ausgearbeitet mit wenig bekannten Straßen, raffiniertem japanischem Essen und einem reichhaltigen Angebot an Kultur und Geschichte. Auf dieser Tour genießt Du einen atemberaubenden Blick auf den Fuji, erlebst die üppigen Wälder der japanischen Alpen und entspannst in den berühmten natürlichen heißen Quellen, die wir auf unserem Weg vorfinden. Erlebe diesen Herbst auf eine ganz besondere andere Art und Weise und komme mit auf diese unglaublich schöne Motorradtour in Japan. Tourdaten Start /Ende: Tokyo Gesamtstrecke: 2.818 km / 1.740 Meilen Dauer: 16 Tage Fahrtage: 14 Tage Rasttage: 3: Kyoto, Hiroshima, Kusatsu Frühstück: 15 Frühstück inbegriffen Abendessen: 12 Abendessen inbegriffen Daily mileage: 160 - 220 km / 250-350 Meilen Hotelübernachtungen: 15 Nätche Fahrsaison: Herbst Higlights: Tokyo, Mount Fuji, Japanese Alps, Tsumago, Hotels: quality hotels and Japanese Ryokans. Ryokans are a Shikoku, Kyoto, Hiroshima, Eiheiji, Matsumoto, Kusatsu, type of traditional Japanese Inn that has existed since the 8th Nikko. century. The oldest hotel in the world is a Japanese Ryokan founded in 718 A.D. Prices Pilot*: 8050€ Passenger: 5800€ Single Room: 1200€ * im Doppelzimmer mit einer Honda NC750X/ SuzukiVStrom650 Bike Upgrade: BMW F750GS + 350€ BMW F850GS/R1200RS + 600€ BMW R1250GS + 895€ BMW R1250RT + 1215€ Tel (USA) (412) 468-2453 [email protected] Tel (España) (+34) 91 633 72 22 www.IMTBike.com Arrive to Japan and transfer to hotel. You will have some free time to relax or explore the Day 1: Odaiba bay area before the welcome briefing. -

CARACTERIZACIÓN GENÉTICA DE RELICTOS DE Guadua Angustifolia, UN ECOSISTEMA ESTRATÉGICO DE LA ECOREGIÓN VALLE DEL CAUCA MEDIANTE STR´S
CARACTERIZACIÓN GENÉTICA DE RELICTOS DE Guadua angustifolia, UN ECOSISTEMA ESTRATÉGICO DE LA ECOREGIÓN VALLE DEL CAUCA MEDIANTE STR´s TESIS DOCTORAL CARLOS ANDRES PEREZ GALINDO Sevilla, 2014 AGRADECIMIENTOS A mi madre Carmenza, abuelos, hermana, sobrino y toda mi familia que siempre ha estado allí, apoyándome en todos los procesos de materialización de mis ideas. Al profesor Heiber Cardenas, que siempre me incentivo a ir más allá en las diferentes investigaciones y en los proyectos que emprendimos, que han estado enmarcados en la construcción de nuevos programas de postgrado en Colombia, estudios en biodiversidad de nuestra gran región Vallecaucana y la puesta en marcha de diferentes proyectos para el mejoramiento formativo en ciencias biológicas de los jóvenes colombianos. A la Universidad Pablo de Olavide por el apoyo no solo académico, sino también económico en mi etapa como becario. A su excelente cuerpo profesoral, que mediante sus enseñanzas me permitieron ampliar no solo mis conocimientos en biotecnología, sino también mi visión sobre la vida, incentivándome a realizar mis estudios de postgrado en bioinformática, lo cual me permitiría tratar de entender la evolución desde la óptica molecular. Al Dr. Modesto Luceño, quien tomaría la tutoría de este importante trabajo doctoral, apoyándome en los diferentes procesos y requerimientos para la presentación de la tesis y comprendiendo las dificultades que se tienen al trabajar a distancia. A la Universidad Santiago de Cali, institución que apoyó económica, logística y moralmente la investigación objeto de este trabajo. Cuyo principal objetivo ha estado centrado en recurrir a la investigación social y científica para la defensa de la biodiversidad de cultivos nativos en una región agroindustrial, regida por los monocultivos de azúcar y que ve como la afectación de la biodiversidad incide de manera negativa en la calidad de las tierras y la disponibilidad de recursos hídricos, lo cual hoy por hoy está afectando a los habitantes de estas zonas. -

Minakami Perfect Travel Guide &
N 4 One Drop Outdoor Guide Service N 9 S 5 English /Enjoy/ /Enjoy/ This popular tour uses inflatable rafts for canoeing around Lake Shima, which is one of the most transparent lakes in Nature the Kanto region. Snow Enjoy activities where you can experience the Enjoy natural snow close to the Tokyo MAP B-3 Wi-Fi richness of nature. Even first-timers can feel at ease metropolitan area. In addition to eight ski resorts, MINAKAMI 0278-72-8136 with rental equipment and experienced guides. 2230-1 Kamimoku, Minakami-machi there is also a variety of other activities. 8:00-18:00 Closed Open daily during the season Perfect Travel Guide & Map onedrop-outdoor.com/ N 1 Minakami Bungy N 5 Norn Minakami Ski Resort S 1 Tanigawadake Tenjindaira Ski Resort Wi-Fi Minakami Mountain Guide Association MAP A-2 This ski resort offers novice to expert Try bungee jumping from the MAP B-3 This ski resort offers gorgeous views of the Tanigawa 0278-72-3575 level trails and Snow Land, an area which Yubiso, Yubukiyama- 42-meter-high Suwakyo Ohashi Bridge mountain range. It is known for its long ski season, Hiking and trekking on Mount Tanigawa, Oze, 0278-62-0401 offers fun in the snow for everyone. Kokuyurin, Minakami-machi over the Tone River. 1744-1 Tsukiyono, lasting from early December to early May. Some areas December to March: and Mount Hotaka are offered here. One of the most Minakami-machi MAP A-3 Wi-Fi have snow as deep as four meters during the high 8:30-16:30 (final lift at popular activities is the eco-hiking tour from Mount 8:30-17:30 16:00), April to end of Tanigawa to Ichinokurasawa, one of Japan's three Closed Open daily during season, offering the ultimate powder skiing without any MAP B-1 0278-72-6688 479-139 Terama, Minakami-machi season: 8:00-17:00 (from greatest rocks, and hiking up Mount Tanigawa with its the season Mid-December to late March: 8:00-16:00 manmade snow. -

List of Volcanoes in Japan
Elevation Elevation Sl. No Name Prefecture Coordinates Last eruption Meter Feet 1 Mount Meakan Hokkaidō 1499 4916 43.38°N 144.02°E 2008 2 Mount Asahi (Daisetsuzan) Hokkaidō 2290 7513 43.661°N 142.858°E 1739 3 Lake Kuttara Hokkaidō 581 1906 42.489°N 141.163°E - 4 Lake Mashū Hokkaidō 855 2805 43.570°N 144.565°E - 5 Nigorigawa Hokkaidō 356 1168 42.12°N 140.45°E Pleistocene 6 Nipesotsu-Maruyama Volcanic Group Hokkaidō 2013 6604 43.453°N 143.036°E 1899 7 Niseko Hokkaidō 1154 3786 42.88°N 140.63°E 4050 BC 8 Oshima Hokkaidō 737 2418 41.50°N 139.37°E 1790 9 Mount Rausu Hokkaidō 1660 5446 44.073°N 145.126°E 1880 10 Mount Rishiri Hokkaidō 1721 5646 45.18°N 141.25°E 5830 BC 11 Shikaribetsu Volcanic Group Hokkaidō 1430 4692 43.312°N 143.096°E Holocene 12 Lake Shikotsu Hokkaidō 1320 4331 42.70°N 141.33°E holocene 13 Mount Shiretoko Hokkaidō 1254 4114 44°14′09″N 145°16′26″E 200000 BC 14 Mount Iō (Shiretoko) Hokkaidō 1563 5128 44.131°N 145.165°E 1936 15 Shiribetsu Hokkaidō 1107 3632 42.767°N 140.916°E Holocene 16 Shōwa-shinzan Hokkaidō 731 2400 42.5°N 140.8°E 1945 17 Mount Yōtei Hokkaidō 1898 6227 42.5°N 140.8°E 1050 BC 18 Abu (volcano) Honshū 571 - 34.50°N 131.60°E - 19 Mount Adatara Honshū 1718 5635 37.62°N 140.28°E 1990 20 Mount Akagi Honshū 1828 5997 36.53°N 139.18°E - 21 Akita-Komaga-Take Honshū 1637 5371 39.75°N 140.80°E 1971 22 Akita-Yake-Yama Honshū 1366 4482 39.97°N 140.77°E 1997 23 Mount Asama Honshū 2544 8340 36.24°N 138.31°E 2009 24 Mount Azuma Honshū 1705 5594 37.73°N 140.25°E 1977 25 Mount Bandai Honshū 1819 5968 37.60°N 140.08°E 1888 -

Molecular Taxonomy of Bambusicolous Fungi: Tetraplosphaeriaceae, a New Pleosporalean Family with Tetraploa-Like Anamorphs
available online at www.studiesinmycology.org StudieS in Mycology 64: 175–209. 2009. doi:10.3114/sim.2009.64.10 Molecular taxonomy of bambusicolous fungi: Tetraplosphaeriaceae, a new pleosporalean family with Tetraploa-like anamorphs K. Tanaka1*, K. Hirayama1, H. Yonezawa1, S. Hatakeyama1, Y. Harada1, T. Sano1, T. Shirouzu2 and T. Hosoya3 1Faculty of Agriculture & Life Sciences, Hirosaki University, Bunkyo-cho 3, Hirosaki, Aomori 036-8561, Japan; 2Fungus/Mushroom Resource and Research Center, Tottori University, Minami 4-101, Koyama, Tottori, Tottori 680-8553 Japan; 3National Museum of Nature and Science, Amakubo 4-1-1, Tsukuba, Ibaraki 305-0005, Japan *Correspondence: Kazuaki Tanaka, [email protected] Abstract: A new pleosporalean family Tetraplosphaeriaceae is established to accommodate five new genera; 1) Tetraplosphaeria with small ascomata and anamorphs belonging to Tetraploa s. str., 2) Triplosphaeria characterised by hemispherical ascomata with rim-like side walls and anamorphs similar to Tetraploa but with three conidial setose appendages, 3) Polyplosphaeria with large ascomata surrounded by brown hyphae and anamorphs producing globose conidia with several setose appendages, 4) Pseudotetraploa, an anamorphic genus, having obpyriform conidia with pseudosepta and four to eight setose appendages, and 5) Quadricrura, an anamorphic genus, having globose conidia with one or two long setose appendages at the apex and four to five short setose appendages at the base. Fifteen new taxa in these genera mostly collected from bamboo are described and illustrated. They are linked by their Tetraploa s. l. anamorphs. To infer phylogenetic placement in the Pleosporales, analyses based on a combined dataset of small- and large-subunit nuclear ribosomal DNA (SSU+LSU nrDNA) was carried out. -

Mt.AKAGI Gunma Prefecture
Maebashi Access MAEBASHI TRAVEL GUIDE & MAP 黒檜山 Fukushima 赤城山第1赤城山第1スキー場 1828 Niigata 津久田駅 P.2・3 Mt. Kurobi 雪山登山 Take Prefecture Prefecture N P.5 Mt. Akagi No. 1 Ski Slope Snowy Mountain By Train 大沼 白樺牧場 Lake Onuma 1685 Climbing P.5 P.5 Shirakaba Farm P.2・4・5 駒ヶ岳 Asakusa Haneda Airport Narita Airport 前 橋 市 Mt. Komagatake Tochigi Station International Terminal Station 県立赤城公園ビジターセンター P.2・3 Free Maebashi City Prefecture 0 2km 1674 Terminal Station 城 P.2 The Akagi Visitors Center 地蔵岳 覚満淵 Mt. Jizodake Kakumanbuchi 赤城温泉ホテル Marsh P.3・5 Airport Terminal Shibukawa P.2 Akagi Onsen Hotel 1579 Keisei Skyliner ¥ 長七郎山 0 Limited Express 2 Station City 花の宿 湯之沢館 Mt. Choshichiro Gunma P.2 Hana no Yado Yunosawakan 1 Approx. 17 353Prefecture 御宿 総本家 25 min. P.2 Onyado Sohonke Keisei Skyliner 1332 滝沢館 Limited Express Saitama 鍋割山 赤城温泉 Takizawakan Shinagawa Nagano Mt. Nabewari Approx. Mito Station Prefecture Akagi Onsen P.2 Station Prefecture 富士見温泉 見晴らしの湯ふれあい館 45 min. 滝沢温泉 Fujimi Spa FUREAIKAN P.3 忠治温泉 Takizawa Onsen 道の駅 ふじみ Chuji Onsen 渋川駅 Road Station Fujimi 旅籠 忠治館 Kiryu JR Yamanote Line Keisei JR Joban Line Shibukawa 353 P.2 Approx. 10 min. Limited Express Station Hatago Chujikan Tobu Railway Ueno Station 赤城神社 City Express Approx. 17 前橋温泉 クア・イ・テルメ Akagi Shrine 赤城南面千本桜 70 min. 桑風庵本店 Approx. Walk Maebashi Spa Sofuan Honten Akagi Cherry 渋川伊香保渋川伊香保 KUR Y THERME P.3 110 min. 5 min. P.7 空っ風家 Blossoms P.4 関 関 Shibukawa Tokyo Station 越 越 P.7 K K Ikaho Karakkaze-ya 自 自 焼肉乃上州敷島店 a a 動 動 n P.6 車 Yakiniku no Joushu Shikishima Branch 赤城高原温泉 Joetsu/Nagano - Ueno Station E 道 353 敷島公園 ばら園 Akagi Kogen Onsen Shinkansen t 17 17 s ぐんまフラワーパーク u Shikishima Koen Rose Garden P.4 Approx. -
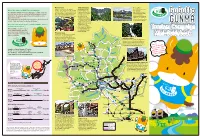
From Narita International Airport from Haneda Airport Transportation Guide There Sa Never-Ending List of Fun, Memorable E
▼ Shima Onsen ▶ Mount Tanigawa ◀ Ozegahara Shima is celebrated as “the spring of Straddling the border Oze is one of the precious What is the GuGutto GUNMA Tourism Campaign? beauties.” This onsen town is full of simple, between Gunma and few remaining nostalgic charm. A famous event known as Niigata prefectures, this is The GuGutto GUNMA Tourism Campaign is a public-relations high-altitude wetlands. It's the “Chochin (Paper Lantern) Walk” is held counted as one of Japan’s a world rich in the variety program designed to spread the word about Gunma’s many here, in which people walk through the hundred most famous of rare living things, attractions, showing people all the excitement there is to town swinging paper lanterns in one hand. mountains. Mount including mountain plants discover in Gunma. Tanigawa’s rock walls, and insects. The grandeur The campaign is packed with new experiences and a variety of including Ichinokurasawa, and refreshing atmosphere are among the three most are a pleasure to witness. events. Come and taste the charms of Gunma! notable rock formations in Japan, and are considered Here’s what’s behind the theme, “GuGutto GUNMA ‒ Discover to be the Mecca of exciting new experiences.”: Japanese rock climbing. From trekking to full-blown We would like everyone to get mountain climbing, there excited with visiting attractive are many ways to enjoy spots and events in Gunma and Mount Tanigawa. ▼ Kusatsu Onsen have memorable experiences that Kusatsu is one of Japan’s most renowned get to your heart. onsen resorts, boasting the highest amount of natural hot water discharge of Oze any onsen resorts in the country.