Nuclear Genes Carbamoyl Phosphate Synthetase And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phylogeny and Biogeography of Hawkmoths (Lepidoptera: Sphingidae): Evidence from Five Nuclear Genes
Phylogeny and Biogeography of Hawkmoths (Lepidoptera: Sphingidae): Evidence from Five Nuclear Genes Akito Y. Kawahara1*, Andre A. Mignault1, Jerome C. Regier2, Ian J. Kitching3, Charles Mitter1 1 Department of Entomology, College Park, Maryland, United States of America, 2 Center for Biosystems Research, University of Maryland Biotechnology Institute, College Park, Maryland, United States of America, 3 Department of Entomology, The Natural History Museum, London, United Kingdom Abstract Background: The 1400 species of hawkmoths (Lepidoptera: Sphingidae) comprise one of most conspicuous and well- studied groups of insects, and provide model systems for diverse biological disciplines. However, a robust phylogenetic framework for the family is currently lacking. Morphology is unable to confidently determine relationships among most groups. As a major step toward understanding relationships of this model group, we have undertaken the first large-scale molecular phylogenetic analysis of hawkmoths representing all subfamilies, tribes and subtribes. Methodology/Principal Findings: The data set consisted of 131 sphingid species and 6793 bp of sequence from five protein-coding nuclear genes. Maximum likelihood and parsimony analyses provided strong support for more than two- thirds of all nodes, including strong signal for or against nearly all of the fifteen current subfamily, tribal and sub-tribal groupings. Monophyly was strongly supported for some of these, including Macroglossinae, Sphinginae, Acherontiini, Ambulycini, Philampelini, Choerocampina, and Hemarina. Other groupings proved para- or polyphyletic, and will need significant redefinition; these include Smerinthinae, Smerinthini, Sphingini, Sphingulini, Dilophonotini, Dilophonotina, Macroglossini, and Macroglossina. The basal divergence, strongly supported, is between Macroglossinae and Smerinthinae+Sphinginae. All genes contribute significantly to the signal from the combined data set, and there is little conflict between genes. -

The Mcguire Center for Lepidoptera and Biodiversity
Supplemental Information All specimens used within this study are housed in: the McGuire Center for Lepidoptera and Biodiversity (MGCL) at the Florida Museum of Natural History, Gainesville, USA (FLMNH); the University of Maryland, College Park, USA (UMD); the Muséum national d’Histoire naturelle in Paris, France (MNHN); and the Australian National Insect Collection in Canberra, Australia (ANIC). Methods DNA extraction protocol of dried museum specimens (detailed instructions) Prior to tissue sampling, dried (pinned or papered) specimens were assigned MGCL barcodes, photographed, and their labels digitized. Abdomens were then removed using sterile forceps, cleaned with 100% ethanol between each sample, and the remaining specimens were returned to their respective trays within the MGCL collections. Abdomens were placed in 1.5 mL microcentrifuge tubes with the apex of the abdomen in the conical end of the tube. For larger abdomens, 5 mL microcentrifuge tubes or larger were utilized. A solution of proteinase K (Qiagen Cat #19133) and genomic lysis buffer (OmniPrep Genomic DNA Extraction Kit) in a 1:50 ratio was added to each abdomen containing tube, sufficient to cover the abdomen (typically either 300 µL or 500 µL) - similar to the concept used in Hundsdoerfer & Kitching (1). Ratios of 1:10 and 1:25 were utilized for low quality or rare specimens. Low quality specimens were defined as having little visible tissue inside of the abdomen, mold/fungi growth, or smell of bacterial decay. Samples were incubated overnight (12-18 hours) in a dry air oven at 56°C. Importantly, we also adjusted the ratio depending on the tissue type, i.e., increasing the ratio for particularly large or egg-containing abdomens. -

British Lepidoptera (/)
British Lepidoptera (/) Home (/) Anatomy (/anatomy.html) FAMILIES 1 (/families-1.html) GELECHIOIDEA (/gelechioidea.html) FAMILIES 3 (/families-3.html) FAMILIES 4 (/families-4.html) NOCTUOIDEA (/noctuoidea.html) BLOG (/blog.html) Glossary (/glossary.html) Family: SPHINGIDAE (3SF 13G 18S) Suborder:Glossata Infraorder:Heteroneura Superfamily:Bombycoidea Refs: Waring & Townsend, Wikipedia, MBGBI9 Proboscis short to very long, unscaled. Antenna ~ 1/2 length of forewing; fasciculate or pectinate in male, simple in female; apex pointed. Labial palps long, 3-segmented. Eye large. Ocelli absent. Forewing long, slender. Hindwing ±triangular. Frenulum and retinaculum usually present but may be reduced. Tegulae large, prominent. Leg spurs variable but always present on midtibia. 1st tarsal segment of mid and hindleg about as long as tibia. Subfamily: Smerinthinae (3G 3S) Tribe: Smerinthini Probably characterised by a short proboscis and reduced or absent frenulum Mimas Smerinthus Laothoe 001 Mimas tiliae (Lime Hawkmoth) 002 Smerinthus ocellata (Eyed Hawkmoth) 003 Laothoe populi (Poplar Hawkmoth) (/002- (/001-mimas-tiliae-lime-hawkmoth.html) smerinthus-ocellata-eyed-hawkmoth.html) (/003-laothoe-populi-poplar-hawkmoth.html) Subfamily: Sphinginae (3G 4S) Rest with wings in tectiform position Tribe: Acherontiini Agrius Acherontia 004 Agrius convolvuli 005 Acherontia atropos (Convolvulus Hawkmoth) (Death's-head Hawkmoth) (/005- (/004-agrius-convolvuli-convolvulus- hawkmoth.html) acherontia-atropos-deaths-head-hawkmoth.html) Tribe: Sphingini Sphinx (2S) -

Clicking Caterpillars: Acoustic Aposematism in Antheraea Polyphemus and Other Bombycoidea Sarah G
993 The Journal of Experimental Biology 210, 993-1005 Published by The Company of Biologists 2007 doi:10.1242/jeb.001990 Clicking caterpillars: acoustic aposematism in Antheraea polyphemus and other Bombycoidea Sarah G. Brown1, George H. Boettner2 and Jayne E. Yack1,* 1Department of Biology, Carleton University, Ottawa, Ontario, K1S 5B6, Canada and 2Plant Soil and Insect Sciences, University of Massachusetts, Amherst, MA 01003, USA *Author for correspondence (e-mail: [email protected]) Accepted 21 November 2006 Summary Acoustic signals produced by caterpillars have been correlated sound production with attack, and an increase documented for over 100 years, but in the majority of in attack rate was positively correlated with the number of cases their significance is unknown. This study is the first signals produced. In addition, sound production typically to experimentally examine the phenomenon of audible preceded or accompanied defensive regurgitation. sound production in larval Lepidoptera, focusing on a Bioassays with invertebrates (ants) and vertebrates (mice) common silkmoth caterpillar, Antheraea polyphemus revealed that the regurgitant is deterrent to would-be (Saturniidae). Larvae produce airborne sounds, predators. Comparative evidence revealed that other resembling ‘clicks’, with their mandibles. Larvae typically Bombycoidea species, including Actias luna (Saturniidae) signal multiple times in quick succession, producing trains and Manduca sexta (Sphingidae), also produce airborne that last over 1·min and include 50–55 clicks. Individual sounds upon attack, and that these sounds precede clicks within a train are on average 24.7·ms in duration, regurgitation. The prevalence and adaptive significance of often consisting of multiple components. Clicks are audible warning sounds in caterpillars is discussed. -

A Survey on Sphingidae (Lepidoptera) Species of South Eastern Turkey
Cumhuriyet Science Journal e-ISSN: 2587-246X Cumhuriyet Sci. J., 41(1) (2020) 319-326 ISSN: 2587-2680 http://dx.doi.org/10.17776/csj.574903 A survey on sphingidae (lepidoptera) species of south eastern Turkey with new distributional records Erdem SEVEN 1 * 1 Department of Gastronomy and Culinary Arts, School of Tourism and Hotel Management, Batman University, 72060, Batman, Turkey. Abstract Article info History: This paper provides comments on the Sphingidae species of south eastern Turkey by the field Received:10.06.2019 surveys are conducted between in 2015-2017. A total of 15 species are determined as a result Accepted:20.12.2019 of the investigations from Batman, Diyarbakır and Mardin provinces. With this study, the Keywords: number of sphinx moths increased to 13 in Batman, 14 in Diyarbakır and 8 in Mardin. Among Fauna, them, 7 species for Batman, 4 species for Diyarbakır and 1 species for Mardin are new record. Hawk moths, For each species, original reference, type locality, material examined, distribution in the world New records, and in Turkey, and larval hostplants are given. Adults figures of Smerinthus kindermanni Sphingidae, Lederer, 1852; Marumba quercus ([Denis & Schiffermüller], 1775); Rethera komarovi Turkey. (Christoph, 1885); Macroglossum stellatarum (Linnaeus, 1758); Hyles euphorbiae (Linnaeus, 1758) and H. livornica (Esper, [1780]) are illustrated. 1. Introduction 18, 22-24]: Acherontia atropos (Linnaeus, 1758); Agrius convolvuli (Linnaeus, 1758); Akbesia davidi (Oberthür, 1884); Clarina kotschyi (Kollar, [1849]); C. The Sphingidae family classified in the Sphingoidea syriaca (Lederer, 1855); Daphnis nerii (Linnaeus, Superfamily and species of the family are generally 1758); Deilephila elpenor (Linnaeus, 1758); D. -

Hawk-Moths, Family Sphingidae and Forewings Browner
Hawk-moths, Family Sphingidae and forewings browner. Wings normally held roof-wise along the body when at rest. Distinctive medium to large moths. Power• Larva green, striped with brown. ful fliers, generally with rather narrow, Habitat More sedentary than above pointed forewings. Most larvae are large, species, living mainly in rough flowery striped, and have a 'horn' at the tail end. places where Privet occurs. Status and distributfon Local in S Convolvulus Hawk-moth Britain, widespread on the Continent. Agrilfs c()llu()lulfli Season 6-7. A strikingly large moth; wingspan up to 12cm. Forewings greyish, marbled; hind• Poplar Hawk-moth wings browner. The abdomen is striped La()th()c l)()fJlfli with red, white and black. The proboscis A medium-sized hawk-moth; wingspan up may be up to 13cm long! to 90mm. Wings greyish to pinkish-brown, Habitat A migrant into N Europe from broadly banded, with a single white mark in the Mediterranean area, which may occur the centre of the forewings. Hindwings wherever there are flowers, especially Petu• orange-red at base, usually concealed, and nia and Nicotiana. Breeds on Convolvulus, but show in front of forewings at rest. Larvae only rarely does so in N Europe. green with yellow stripes. Status and distribution Very variable in Habitat A variety of habitats, associated numbers, regularly reaching S England, but with Sallow, Poplar and Aspen. not necessarily going further. Status and distribution Widely distrib• Season 6-9. uted and moderately common throughout the region. Death's Head Hawk-Moth Season 5-9. Achcrontia atrofJos Similar species An extraordinary insect, unlike anything Pine Hawk-moth Hyloicus pinostri is also else. -
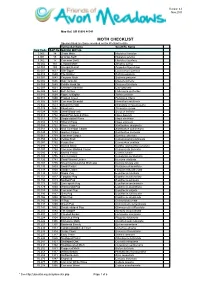
MOTH CHECKLIST Species Listed Are Those Recorded on the Wetland to Date
Version 4.0 Nov 2015 Map Ref: SO 95086 46541 MOTH CHECKLIST Species listed are those recorded on the Wetland to date. Vernacular Name Scientific Name New Code B&F No. MACRO MOTHS 3.005 14 Ghost Moth Hepialus humulae 3.001 15 Orange Swift Hepialus sylvina 3.002 17 Common Swift Hepialus lupulinus 50.002 161 Leopard Moth Zeuzera pyrina 54.008 169 Six-spot Burnet Zygaeba filipendulae 66.007 1637 Oak Eggar Lasiocampa quercus 66.010 1640 The Drinker Euthrix potatoria 68.001 1643 Emperor Moth Saturnia pavonia 65.002 1646 Oak Hook-tip Drepana binaria 65.005 1648 Pebble Hook-tip Drepana falcataria 65.007 1651 Chinese Character Cilix glaucata 65.009 1653 Buff Arches Habrosyne pyritoides 65.010 1654 Figure of Eighty Tethia ocularis 65.015 1660 Frosted Green Polyploca ridens 70.305 1669 Common Emerald Hermithea aestivaria 70.302 1673 Small Emerald Hemistola chrysoprasaria 70.029 1682 Blood-vein Timandra comae 70.024 1690 Small Blood-vein Scopula imitaria 70.013 1702 Small Fan-footed Wave Idaea biselata 70.011 1708 Single-dotted Wave Idaea dimidiata 70.016 1713 Riband Wave Idaea aversata 70.053 1722 Flame Carpet Xanthorhoe designata 70.051 1724 Red Twin-spot Carpet Xanthorhoe spadicearia 70.049 1728 Garden Carpet Xanthorhoe fluctuata 70.061 1738 Common Carpet Epirrhoe alternata 70.059 1742 Yellow Shell Camptogramma bilineata 70.087 1752 Purple Bar Cosmorhoe ocellata 70.093 1758 Barred Straw Eulithis (Gandaritis) pyraliata 70.097 1764 Common Marbled Carpet Chloroclysta truncata 70.085 1765 Barred Yellow Cidaria fulvata 70.100 1776 Green Carpet Colostygia pectinataria 70.126 1781 Small Waved Umber Horisme vitalbata 70.107 1795 November/Autumnal Moth agg Epirrita dilutata agg. -

Results of a Lepidopterological Expedition to North and Northwest Iran in Summer 2007 with New Records for Iran (Lepidoptera) (Plates 19-22)
Esperiana Band 16: 135-165 Schwanfeld, 06. Dezember 2011 ISBN 978-3-938249-01-7 Results of a lepidopterological expedition to North and Northwest Iran in summer 2007 with new records for Iran (Lepidoptera) (plates 19-22) Lutz LEHMANN † & Reza ZAHIRI Abstract: The results of a joint lepidopterological expedition to North and Northwest Iran, povinces of Tehran, Mazandaran, Guilan, Ardabil and Azerbaijan-e-Sharqi, from 20 July to 1 August are presented. More than 508 species of Macrolepidoptera (sensu SEITZ) could be recorded, among them Idaea sericeata (HÜBNER, 1813), Cinglis humifusaria EVERSMANN, 1837, Aplocera uniformata (URBAHN, 1971), Scotopteryx chenopodiata (LINNAEUS, 1758), Triphosa dubitata (LINANEUS, 1758), Abraxas grossulariata (LINNAEUS, 1758), Kemtrognophos ciscaucasica (RJABOV, 1964), Furcula danieli SCHINTLMEISTER, 1998, Eilema lurideola ([ZINCKEN], 1817), Zethes propinquus CHRISTOPH, 1885, Meganola togatulalis (HÜBNER, 1796), Meganola kolbi (DANIEL, 1935), Pseudluperina pozzii (CURÓ, 1883), Mythimna sicula scirpi (DUPONCHEL, 1836) and Noctua interposita (HÜBNER, 1790) new for the fauna of Iran. Additionally, the species of two light traps in the Elburs Mts., province Mazandaran, from the beginning of June, collected by A. PÜTZ, are listed. The genitalia of 16 species, typical habitats, and some living and spread specimens are figured. Zusammenfassung: Die Ergebnisse einer gemeinsamen lepidopterologischen Expedition in den Nord- und Nordwestiran, Provinzen Teheran, Mazandaran, Guilan, Ardabil and Azerbaijan-e-Sharqi, vom -

Biosystems ISSN 2520-2529 (Online) Biosyst
ISSN 2519-8513 (Print) Biosystems ISSN 2520-2529 (Online) Biosyst. Divers., 2020, 28(3), 290–297 Diversity doi: 10.15421/012038 12B Fruit and berry plants of forest belts as a factor of species diversity of ornithofauna during the breeding season and autumn migration period V. V. Pesotskaya*, A. B. Chaplygina*, T. V. Shupova**, R. I. Kratenko* *G. S. Skovoroda Kharkiv National Pedagogical University, Kharkiv, Ukraine **Institute for Evolutionary Ecology of NAS of Ukraine, Kyiv, Ukraine Article info Pesotskaya, V. V., Chaplygina, A. B., Shupova, T. V., & Kratenko, R. I. (2020). Fruit and berry plants of forest belts as a factor of Received 29.07.2020 species diversity of ornithofauna during the breeding season and autumn migration period. Biosystems Diversity, 28(3), 290–297. Received in revised form 02.09.2020 doi:10.15421/012038 Accepted 03.09.2020 During migration, the availability of food that affects the success of bird movements, the nature and timing of their movements, H. S. Skovoroda Kharkiv is critical for many bird species. The relationship between migration routes and the ripening of fruit and berry plants along the route is National Pedagogical University, important. Four types of forest belts were studied: wind-blown maple-ash, latticed maple-linden, dense oak-maple-linden, wind- Alchevsky st., 29, Kharkiv, blown oak-maple-poplar. During the study 43 bird species were identified consuming 9 major fruit and berry plant species: 61002, Ukraine. Sambucus nigra, Prunus spinosa, Crataegus laevigata, Rosa canina, Prunus padus, Sorbus aucuparia, Rhamnus cathartica, Morus Tel.: +38-050-882-50-86. E-mail: [email protected] nigra, Prunus cerasus. -

Evidence for Breeding Lime Hawkmoth (Mimas Tiliae) in Glasgow, Scotland
The Glasgow Naturalist (online 2020) Volume 27, Part 2 https://doi.org/10.37208/tgn27217 surprise, therefore, when a mating pair of lime hawkmoths was photographed in Glebe Street, Renfrew, Evidence for breeding lime Renfrewshire on 27th May 2016. Whilst publicised at the time on the Paisley Natural History Society website hawkmoth (Mimas tiliae) in Glasgow, (PNHS, 2020) and in the local press (Paisley Daily Scotland Express, 20th June 2016), there were no subsequent sightings. Indeed, the possibility was raised that the pair A.P. Payne could have been purchased via one of several commercial operators dealing in hawkmoths. Thomson Building, University of Glasgow, Glasgow G12 8QQ On the late afternoon of 12th August 2019 I found a large green hawkmoth caterpillar on top of a low wall in E-mail: [email protected] Orleans Avenue, Jordanhill, Glasgow (Fig. 1). Close examination revealed a blue horn on the tail with a pink blush on the undersurface of the base. The tail also bore prominent yellow warts surrounding a dark red patch. The lime hawkmoth (Mimas tiliae) is an attractive olive The spiracles were ringed in red and there were seven and sand-coloured, medium-sized hawkmoth with a oblique yellow stripes on the light green body. This was wide distribution in Europe, the Middle East and Asia subsequently confirmed as a final instar lime hawkmoth (Pittaway, 1993). There are two 19th century records of larva. the moth in the Glasgow area: one by John Dunsmore near Houston, Renfrewshire; the other by Robert The caterpillar was found immediately underneath a Dunlop near Kilmarnock, East Ayrshire. -

Lepidoptera: Sphingidae)
Pacific Insects Vol. 23, no. 1-2: 207-210 23 June 1981 © 1981 by the Bishop Museum TRANSFER OF THE SPHINGID GENUS SATASPES FROM THE SUBFAMILY MACROGLOSSINAE TO THE SUBFAMILY SPHINGINAE (LEPIDOPTERA: SPHINGIDAE) By J. C. E. Riotte1 Abstract. The sphingid genus Sataspes is transferred from the subfamily Macroglossinae to the subfamily Sphinginae based on characters of the adult labial palpus and characters of the larva. In their revision of the Sphingidae, Rothschild & Jordan (1903) based their higher classification on a certain character of the labial palpus in the adults and the shape of the head of the larva. They separated the family into 2 groups: "Sphingidae asemanophorae" and "Sphingidae semanophorae." Hodges (1971) erected the subfamilies Sphinginae (for the former) and Macroglossinae (for the latter). As a main character of the labial palpus of the "semanophorae," Rothschild & Jordan (p. 347) established: "The not-scaled area of the inner surface of the first segment of the palpus covered with short sensory hairs, or these hairs, which are seldom vestigial, restricted to a patch." As a decisive character for the larvae they established: "The larvae are not granulose as in Ambulicinae, nor have they ever a triangular head; they are also not regularly banded as in most Protoparce, Hyloicus ligustri, etc." In the genus Sataspes Moore, 1857 the labial palpus has no sensory hairs whatsoever and the head of the larva is triangular. Also, the larva is granulose and regularly striped and tapers off cephalad as in Mimas. Mell (1922), in his exhaustive work on the S China Sphingidae, described for the first time and figured in color larvae of Sataspes. -

The Spanish Pyrenees
The Spanish Pyrenees Naturetrek Tour Report 13 - 20 May 2012 Giant Peacock courtesy of Tim Crafer Group photo by Janet Blizard Berdun by Janet Blizard Report compiled by Philip Thompson Naturetrek Cheriton Mill Cheriton Alresford Hampshire SO24 0NG England T: +44 (0)1962 733051 F: +44 (0)1962 736426 E: [email protected] W: www.naturetrek.co.uk Tour REport The Spanish Pyrenees Tour Leaders:- Philip Thompson Byron Palacios Participants:- Tim Crafer Shelagh Crafer Liz Savory Alan Dawson Chris Dawson Annie Green Steve Tilbury Kate Tilbury Tim Lait Rita Lait Janet Blizard Louise Thompson Day 1 Sunday 13th May Once the group had assembled we were quickly away in our tour minibuses in the early evening heading north across the Zaragoza plains towards the Pyrenees. Several Black Kites were seen as we passed, followed by a flock of Cattle Egrets and single Marsh Harrier at a small wetland beside the motorway. We enjoyed a lovely sunset as we climbed through the pre-Pyrenean hills and then dropped down into La Canal de Berdun and got our first view of Berdun itself perched on its isolated hilltop. We enjoyed a late meal and refreshments before retiring to bed. Day 2 Monday 14th May After our long day of travel the previous day we enjoyed a pleasant walk into the ‘Badlands’ of eroded slopes below the village of Berdun down towards the Rio Veral. As we set off from the village several wayside plants were picked out which included both Erodium malacoides and ciconium and Papaver hybridum. A very obliging Nightingale sat out in the open for some time allowing everyone to get a great view of these usually very secretive and skulking birds which nonetheless are generally very common and heard singing throughout the trip.