Supporting Information
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -
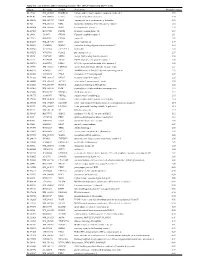
Supplementary Table 1
Table S1. List of Genes Differentially Expressed in YB-1 siRNA-Transfected MCF-7 Cells Unigene Accession Symbol Description Mean fold change Hs.17466 NM_004585 RARRES3 retinoic acid receptor responder (tazarotene induced) 3 3.57 Hs.64746 NM_004669 CLIC3 chloride intracellular channel 3 3.33 Hs.516155 NM_001747 CAPG capping protein (actin filament), gelsolin-like 2.88 Hs.926 NM_002463 MX2 myxovirus (influenza virus) resistance 2 (mouse) 2.72 Hs.643494 NM_005410 SEPP1 selenoprotein P, plasma, 1 2.70 Hs.267038 BC017500 POF1B premature ovarian failure 1B 2.67 Hs.20961 U63917 GPR30 G protein- coupled receptor 30 2532.53 Hs.199877 BE645967 CPNE4 copine IV 2.49 Hs.632177 NM_017458 MVP major vault protein 2.48 Hs.528836 AA005023 NOD27 nucleotide-binding oligomerization domains 27 2.43 Hs.360940 AL589866 dJ222E13.1 kraken-like 2.40 Hs.515575 AI743780 PLAC2 placenta-specific 2 2.37 Hs.25674 AI827820 MBD2 methyl-CpG binding domain protein 2 2.36 Hs.12229 AA149594 TIEG2 TGFB inducible early growth response 2 2.33 Hs.482730 AA053711 EDIL3 EGF-like repeats and discoidin I-like domains 3 2.32 Hs. 370771 NM_000389 CDKN1A cyclin- depen dent kinase in hibitor 1A (p 21, Cip 1) 2282.28 Hs.469175 AI341537 JFC1 NADPH oxidase-related, C2 domain-containing protein 2.28 Hs.514821 AF043341 CCL5 chemokine (C-C motif) ligand 5 2.27 Hs.591292 NM_023915 GPR87 G protein-coupled receptor 87 2.26 Hs.500483 NM_001613 ACTA2 actin, alpha 2, smooth muscle, aorta 2.24 Hs.632824 NM_006729 DIAPH2 diaphanous homolog 2 (Drosophila) 2.22 Hs.369430 NM_000919 PAM peptidylglycine -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Phosphoproteomics of Retinoblastoma: a Pilot Study Identifies Aberrant Kinases
molecules Article Phosphoproteomics of Retinoblastoma: A Pilot Study Identifies Aberrant Kinases Lakshmi Dhevi Nagarajha Selvan 1,†, Ravikanth Danda 1,2,†, Anil K. Madugundu 3 ID , Vinuth N. Puttamallesh 3, Gajanan J. Sathe 3,4, Uma Maheswari Krishnan 2, Vikas Khetan 5, Pukhraj Rishi 5, Thottethodi Subrahmanya Keshava Prasad 3,6 ID , Akhilesh Pandey 3,7,8, Subramanian Krishnakumar 1, Harsha Gowda 3,* and Sailaja V. Elchuri 9,* 1 L&T Opthalmic Pathology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (L.D.N.S.); [email protected] (R.D.); [email protected] (S.K.) 2 Centre for Nanotechnology and Advanced Biomaterials, Shanmugha Arts, Science, Technology and Research Academy University, Tanjore, Tamil Nadu 613 401, India; [email protected] 3 Institute of Bioinformatics, International Technology Park, Bangalore, Karnataka 560 066, India; [email protected] (A.K.M.); [email protected] (V.N.P.); [email protected] (G.J.S.); [email protected] (T.S.K.P.); [email protected] (A.P.) 4 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576 104, India 5 Shri Bhagwan Mahavir Vitreoretinal Services, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India; [email protected] (V.K.); [email protected] (P.R.) 6 Center for Systems Biology and Molecular Medicine, Yenepoya Research Centre, Yenepoya (Deemed to be University), Mangalore, Karnataka 575 108, India 7 McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 8 Departments of Biological Chemistry, Pathology and Oncology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA 9 Department of Nanotechnology, Vision Research Foundation, Sankara Nethralaya, Chennai, Tamil Nadu 600 006, India * Correspondence: [email protected] (H.G.); [email protected] (S.V.E.); Tel.: +91-80-28416140 (H.G.); +91-44-28271616 (S.V.E.) † These authors contributed equally to this work. -

PHKB Gene Phosphorylase Kinase Regulatory Subunit Beta
PHKB gene phosphorylase kinase regulatory subunit beta Normal Function The PHKB gene provides instructions for making one piece, the beta subunit, of the phosphorylase b kinase enzyme. This enzyme is made up of 16 subunits, four each of the alpha, beta, gamma, and delta subunits. (Each subunit is produced from a different gene.) The beta subunit helps regulate the activity of phosphorylase b kinase. This enzyme is found in various tissues, although it is most abundant in the liver and muscles. One version of the enzyme is found in liver cells and another in muscle cells. The beta subunit produced from the PHKB gene is part of the enzyme found both in the liver and in muscle. Phosphorylase b kinase plays an important role in providing energy for cells. The main source of cellular energy is a simple sugar called glucose. Glucose is stored in muscle and liver cells in a form called glycogen. Glycogen can be broken down rapidly when glucose is needed, for instance during exercise. Phosphorylase b kinase turns on ( activates) another enzyme called glycogen phosphorylase b by converting it to the more active form, glycogen phosphorylase a. When active, this enzyme breaks down glycogen. Health Conditions Related to Genetic Changes Glycogen storage disease type IX At least 21 mutations in the PHKB gene are known to cause a form of glycogen storage disease type IX (GSD IX) called GSD IXb. This form of the disorder affects the liver and the muscles. The liver problems caused by this disorder include an enlarged liver ( hepatomegaly), slow growth, and periods of low blood sugar (hypoglycemia). -

Clinical, Biochemical, and Genetic Characterization of Glycogen Storage Type IX in a Child with Asymptomatic Hepatomegaly
pISSN: 2234-8646 eISSN: 2234-8840 http://dx.doi.org/10.5223/pghn.2015.18.2.138 Pediatr Gastroenterol Hepatol Nutr 2015 June 18(2):138-143 Case Report PGHN Clinical, Biochemical, and Genetic Characterization of Glycogen Storage Type IX in a Child with Asymptomatic Hepatomegaly Jung Ah Kim, Ja Hye Kim, Beom Hee Lee, Gu-Hwan Kim*, Yoon S. Shin†, Han-Wook Yoo, and Kyung Mo Kim Department of Pediatrics, *Medical Genetics Center, Asan Medical Center Children’s Hospital, University of Ulsan College of Medicine, Seoul, Korea, †University Children's Hospital and Molecular Genetics and Metabolism Laboratory, Munich, Germany Glycogen storage disease type IX (GSD IX) is caused by a defect in phosphorylase b kinase (PhK) that results from mutations in the PHKA2, PHKB, and PHKG2 genes. Patients usually manifest recurrent ketotic hypoglycemia with growth delay, but some may present simple hepatomegaly. Although GSD IX is one of the most common causes of GSDs, its biochemical and genetic diagnosis has been problematic due to its rarity, phenotypic overlap with other types of GSDs, and genetic heterogeneities. In our report, a 22-month-old boy with GSD IX is described. No other manifestations were evident except for hepatomegaly. His growth and development also have been proceeding normally. Diagnosed was made by histologic examination, an enzyme assay, and genetic testing with known c.3210_3212del (p.Arg1070del) mutation in PHKA2 gene. Key Words: Glycogen storage disease, Glycogen storage disease type IX, Phosphorylase b kinase 2, Phosphorylase kinase, Hepatomegaly INTRODUCTION lism [1]. GSD type IX (GSD IX) results from a defi- ciency of phosphorylase b kinase (PhK), which plays Glycogen storage disease (GSD) is a group of dis- an essential role in regulating the breakdown of gly- eases caused by inborn errors of glycogen metabo- cogen to glucose. -

Download 20190410); Fragmentation for 20 S
ARTICLE https://doi.org/10.1038/s41467-020-17387-y OPEN Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes ✉ Martin Mehnert 1 , Rodolfo Ciuffa1, Fabian Frommelt 1, Federico Uliana1, Audrey van Drogen1, ✉ ✉ Kilian Ruminski1,3, Matthias Gstaiger1 & Ruedi Aebersold 1,2 fi 1234567890():,; Rapidly increasing availability of genomic data and ensuing identi cation of disease asso- ciated mutations allows for an unbiased insight into genetic drivers of disease development. However, determination of molecular mechanisms by which individual genomic changes affect biochemical processes remains a major challenge. Here, we develop a multilayered proteomic workflow to explore how genetic lesions modulate the proteome and are trans- lated into molecular phenotypes. Using this workflow we determine how expression of a panel of disease-associated mutations in the Dyrk2 protein kinase alter the composition, topology and activity of this kinase complex as well as the phosphoproteomic state of the cell. The data show that altered protein-protein interactions caused by the mutations are asso- ciated with topological changes and affected phosphorylation of known cancer driver pro- teins, thus linking Dyrk2 mutations with cancer-related biochemical processes. Overall, we discover multiple mutation-specific functionally relevant changes, thus highlighting the extensive plasticity of molecular responses to genetic lesions. 1 Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, -

The Mitotic Checkpoint Is a Targetable Vulnerability of Carboplatin-Resistant
www.nature.com/scientificreports OPEN The mitotic checkpoint is a targetable vulnerability of carboplatin‑resistant triple negative breast cancers Stijn Moens1,2, Peihua Zhao1,2, Maria Francesca Baietti1,2, Oliviero Marinelli2,3, Delphi Van Haver4,5,6, Francis Impens4,5,6, Giuseppe Floris7,8, Elisabetta Marangoni9, Patrick Neven2,10, Daniela Annibali2,11,13, Anna A. Sablina1,2,13 & Frédéric Amant2,10,12,13* Triple‑negative breast cancer (TNBC) is the most aggressive breast cancer subtype, lacking efective therapy. Many TNBCs show remarkable response to carboplatin‑based chemotherapy, but often develop resistance over time. With increasing use of carboplatin in the clinic, there is a pressing need to identify vulnerabilities of carboplatin‑resistant tumors. In this study, we generated carboplatin‑resistant TNBC MDA‑MB‑468 cell line and patient derived TNBC xenograft models. Mass spectrometry‑based proteome profling demonstrated that carboplatin resistance in TNBC is linked to drastic metabolism rewiring and upregulation of anti‑oxidative response that supports cell replication by maintaining low levels of DNA damage in the presence of carboplatin. Carboplatin‑ resistant cells also exhibited dysregulation of the mitotic checkpoint. A kinome shRNA screen revealed that carboplatin‑resistant cells are vulnerable to the depletion of the mitotic checkpoint regulators, whereas the checkpoint kinases CHEK1 and WEE1 are indispensable for the survival of carboplatin‑ resistant cells in the presence of carboplatin. We confrmed that pharmacological inhibition of CHEK1 by prexasertib in the presence of carboplatin is well tolerated by mice and suppresses the growth of carboplatin‑resistant TNBC xenografts. Thus, abrogation of the mitotic checkpoint by CHEK1 inhibition re‑sensitizes carboplatin‑resistant TNBCs to carboplatin and represents a potential strategy for the treatment of carboplatin‑resistant TNBCs. -

Human Kinases Info Page
Human Kinase Open Reading Frame Collecon Description: The Center for Cancer Systems Biology (Dana Farber Cancer Institute)- Broad Institute of Harvard and MIT Human Kinase ORF collection from Addgene consists of 559 distinct human kinases and kinase-related protein ORFs in pDONR-223 Gateway® Entry vectors. All clones are clonal isolates and have been end-read sequenced to confirm identity. Kinase ORFs were assembled from a number of sources; 56% were isolated as single cloned isolates from the ORFeome 5.1 collection (horfdb.dfci.harvard.edu); 31% were cloned from normal human tissue RNA (Ambion) by reverse transcription and subsequent PCR amplification adding Gateway® sequences; 11% were cloned into Entry vectors from templates provided by the Harvard Institute of Proteomics (HIP); 2% additional kinases were cloned into Entry vectors from templates obtained from collaborating laboratories. All ORFs are open (stop codons removed) except for 5 (MST1R, PTK7, JAK3, AXL, TIE1) which are closed (have stop codons). Detailed information can be found at: www.addgene.org/human_kinases Handling and Storage: Store glycerol stocks at -80oC and minimize freeze-thaw cycles. To access a plasmid, keep the plate on dry ice to prevent thawing. Using a sterile pipette tip, puncture the seal above an individual well and spread a portion of the glycerol stock onto an agar plate. To patch the hole, use sterile tape or a portion of a fresh aluminum seal. Note: These plasmid constructs are being distributed to non-profit institutions for the purpose of basic -

Mutations in PHKA1, PHKG1 Or Six Other Candidate Genes Explain Only a Minority of Cases
European Journal of Human Genetics (2003) 11, 516–526 & 2003 Nature Publishing Group All rights reserved 1018-4813/03 $25.00 www.nature.com/ejhg ARTICLE Muscle glycogenosis with low phosphorylase kinase activity: mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases Barbara Burwinkel1,7, Bin Hu1, Anja Schroers2, Paula R. Clemens3,8, Shimon W. Moses4, Yoon S. Shin5, Dieter Pongratz6, Matthias Vorgerd2 and Manfred W. Kilimann*,1,9 1Institut fu¨r Physiologische Chemie, Ruhr-Universita¨t Bochum, D-44780 Bochum, Germany; 2Neurologische Universita¨tsklinik Bergmannsheil, Ruhr-Universita¨t Bochum, Bu¨rkle-de-la-Camp-Platz 1, D-44789 Bochum, Germany; 3Department of Neurology, Mayo Clinic, Rochester, MN 55905, USA; 4Department of Pediatrics, Soroka Medical Center, Ben Gurion University of the Negev, IL-84105 Beer-Sheva, Israel; 5Stoffwechselzentrum, Dr V Haunersches Kinderspital der Universita¨t Mu¨nchen, D-80337 Mu¨nchen, Germany; 6Friedrich-Baur-Institut der Universita¨t Mu¨nchen, Ziemssenstr. 1, D-80336 Mu¨nchen, Germany Muscle-specific deficiency of phosphorylase kinase (Phk) causes glycogen storage disease, clinically manifesting in exercise intolerance with early fatiguability, pain, cramps and occasionally myoglobinuria. In two patients and in a mouse mutant with muscle Phk deficiency, mutations were previously found in the muscle isoform of the Phk a subunit, encoded by the X-chromosomal PHKA1 gene (MIM # 311870). No mutations have been identified in the muscle isoform of the Phk c subunit (PHKG1). In the present study, we determined the structure of the PHKG1 gene and characterized its relationship to several pseudogenes. In six patients with adult- or juvenile-onset muscle glycogenosis and low Phk activity, we then searched for mutations in eight candidate genes. -

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

Cell Culture-Based Profiling Across Mammals Reveals DNA Repair And
1 Cell culture-based profiling across mammals reveals 2 DNA repair and metabolism as determinants of 3 species longevity 4 5 Siming Ma1, Akhil Upneja1, Andrzej Galecki2,3, Yi-Miau Tsai2, Charles F. Burant4, Sasha 6 Raskind4, Quanwei Zhang5, Zhengdong D. Zhang5, Andrei Seluanov6, Vera Gorbunova6, 7 Clary B. Clish7, Richard A. Miller2, Vadim N. Gladyshev1* 8 9 1 Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard 10 Medical School, Boston, MA, 02115, USA 11 2 Department of Pathology and Geriatrics Center, University of Michigan Medical School, 12 Ann Arbor, MI 48109, USA 13 3 Department of Biostatistics, School of Public Health, University of Michigan, Ann Arbor, 14 MI 48109, USA 15 4 Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI 16 48109, USA 17 5 Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10128, USA 18 6 Department of Biology, University of Rochester, Rochester, NY 14627, USA 19 7 Broad Institute, Cambridge, MA 02142, US 20 21 * corresponding author: Vadim N. Gladyshev ([email protected]) 22 ABSTRACT 23 Mammalian lifespan differs by >100-fold, but the mechanisms associated with such 24 longevity differences are not understood. Here, we conducted a study on primary skin 25 fibroblasts isolated from 16 species of mammals and maintained under identical cell culture 26 conditions. We developed a pipeline for obtaining species-specific ortholog sequences, 27 profiled gene expression by RNA-seq and small molecules by metabolite profiling, and 28 identified genes and metabolites correlating with species longevity. Cells from longer-lived 29 species up-regulated genes involved in DNA repair and glucose metabolism, down-regulated 30 proteolysis and protein transport, and showed high levels of amino acids but low levels of 31 lysophosphatidylcholine and lysophosphatidylethanolamine.