Mutations in PHKA1, PHKG1 Or Six Other Candidate Genes Explain Only a Minority of Cases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phosphorylation of Mcardle Phosphorylase Induces Activity (Human Skeletal Muscle/Protein Kinase) CESARE G
Proc. Nati. Acad. Sci. USA Vol. 78, No. 5, pp. 2688-2692, May 1981 Biochemistry Phosphorylation of McArdle phosphorylase induces activity (human skeletal muscle/protein kinase) CESARE G. CERRI AND JOSEPH H. WILLNER Department of Neurology and H. Houston Merritt Clinical Research Center for Muscular Dystrophy and Related Diseases, Columbia University College of Physicians and Surgeons, New York, New York 10032 Communicated by Harry Grundfest, January 7, 1981 ABSTRACT In McArdle disease, myophosphorylase defi- mediate between those of phosphorylases b and a. Karpatkin ciency, enzyme activity is absent but the presence of an altered et al. (19, 20) found that incubation of human platelets with enzyme protein can frequently be demonstrated. We have found MgATP+ resulted in an increase in total phosphorylase activity that phosphorylation of this protein in vitro can result in catalytic and concluded that the data were "consistent with the presence activity. We studied muscle of four patients; all lacked myophos- in human platelets of inactive dimer and monomer species of phorylase activity, but myophosphorylase protein was demon- phosphorylase, which require MgATP for activation." Because strated by immunodiffusion or gel electrophoresis. Incubation of activation of these isozymes was probably due to protein phos- muscle homogenate supernatants with cyclic AMP-dependent pro- phorylation and also because incomplete phosphorylation could tein kinase and ATP resulted in phosphorylase activity. The ac- tivated enzyme comigrated with normal human myophosphory- result in reduced activity, we evaluated the possibility that the lase in gel electrophoresis. Incubation with [y-32P]ATP resulted activity ofphosphorylase in McArdle muscle could be restored in incorporation of 32P into the band possessing phosphorylase by phosphorylation of the inactive phosphorylase protein pres- activity. -

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

Gene Symbol Gene Description ACVR1B Activin a Receptor, Type IB
Table S1. Kinase clones included in human kinase cDNA library for yeast two-hybrid screening Gene Symbol Gene Description ACVR1B activin A receptor, type IB ADCK2 aarF domain containing kinase 2 ADCK4 aarF domain containing kinase 4 AGK multiple substrate lipid kinase;MULK AK1 adenylate kinase 1 AK3 adenylate kinase 3 like 1 AK3L1 adenylate kinase 3 ALDH18A1 aldehyde dehydrogenase 18 family, member A1;ALDH18A1 ALK anaplastic lymphoma kinase (Ki-1) ALPK1 alpha-kinase 1 ALPK2 alpha-kinase 2 AMHR2 anti-Mullerian hormone receptor, type II ARAF v-raf murine sarcoma 3611 viral oncogene homolog 1 ARSG arylsulfatase G;ARSG AURKB aurora kinase B AURKC aurora kinase C BCKDK branched chain alpha-ketoacid dehydrogenase kinase BMPR1A bone morphogenetic protein receptor, type IA BMPR2 bone morphogenetic protein receptor, type II (serine/threonine kinase) BRAF v-raf murine sarcoma viral oncogene homolog B1 BRD3 bromodomain containing 3 BRD4 bromodomain containing 4 BTK Bruton agammaglobulinemia tyrosine kinase BUB1 BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) BUB1B BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) C9orf98 chromosome 9 open reading frame 98;C9orf98 CABC1 chaperone, ABC1 activity of bc1 complex like (S. pombe) CALM1 calmodulin 1 (phosphorylase kinase, delta) CALM2 calmodulin 2 (phosphorylase kinase, delta) CALM3 calmodulin 3 (phosphorylase kinase, delta) CAMK1 calcium/calmodulin-dependent protein kinase I CAMK2A calcium/calmodulin-dependent protein kinase (CaM kinase) II alpha CAMK2B calcium/calmodulin-dependent -

Enzymatic Encoding Methods for Efficient Synthesis Of
(19) TZZ__T (11) EP 1 957 644 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C12N 15/10 (2006.01) C12Q 1/68 (2006.01) 01.12.2010 Bulletin 2010/48 C40B 40/06 (2006.01) C40B 50/06 (2006.01) (21) Application number: 06818144.5 (86) International application number: PCT/DK2006/000685 (22) Date of filing: 01.12.2006 (87) International publication number: WO 2007/062664 (07.06.2007 Gazette 2007/23) (54) ENZYMATIC ENCODING METHODS FOR EFFICIENT SYNTHESIS OF LARGE LIBRARIES ENZYMVERMITTELNDE KODIERUNGSMETHODEN FÜR EINE EFFIZIENTE SYNTHESE VON GROSSEN BIBLIOTHEKEN PROCEDES DE CODAGE ENZYMATIQUE DESTINES A LA SYNTHESE EFFICACE DE BIBLIOTHEQUES IMPORTANTES (84) Designated Contracting States: • GOLDBECH, Anne AT BE BG CH CY CZ DE DK EE ES FI FR GB GR DK-2200 Copenhagen N (DK) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI • DE LEON, Daen SK TR DK-2300 Copenhagen S (DK) Designated Extension States: • KALDOR, Ditte Kievsmose AL BA HR MK RS DK-2880 Bagsvaerd (DK) • SLØK, Frank Abilgaard (30) Priority: 01.12.2005 DK 200501704 DK-3450 Allerød (DK) 02.12.2005 US 741490 P • HUSEMOEN, Birgitte Nystrup DK-2500 Valby (DK) (43) Date of publication of application: • DOLBERG, Johannes 20.08.2008 Bulletin 2008/34 DK-1674 Copenhagen V (DK) • JENSEN, Kim Birkebæk (73) Proprietor: Nuevolution A/S DK-2610 Rødovre (DK) 2100 Copenhagen 0 (DK) • PETERSEN, Lene DK-2100 Copenhagen Ø (DK) (72) Inventors: • NØRREGAARD-MADSEN, Mads • FRANCH, Thomas DK-3460 Birkerød (DK) DK-3070 Snekkersten (DK) • GODSKESEN, -
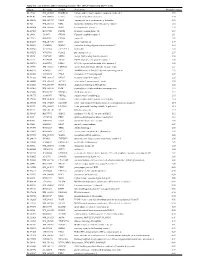
Supplementary Table 1
Table S1. List of Genes Differentially Expressed in YB-1 siRNA-Transfected MCF-7 Cells Unigene Accession Symbol Description Mean fold change Hs.17466 NM_004585 RARRES3 retinoic acid receptor responder (tazarotene induced) 3 3.57 Hs.64746 NM_004669 CLIC3 chloride intracellular channel 3 3.33 Hs.516155 NM_001747 CAPG capping protein (actin filament), gelsolin-like 2.88 Hs.926 NM_002463 MX2 myxovirus (influenza virus) resistance 2 (mouse) 2.72 Hs.643494 NM_005410 SEPP1 selenoprotein P, plasma, 1 2.70 Hs.267038 BC017500 POF1B premature ovarian failure 1B 2.67 Hs.20961 U63917 GPR30 G protein- coupled receptor 30 2532.53 Hs.199877 BE645967 CPNE4 copine IV 2.49 Hs.632177 NM_017458 MVP major vault protein 2.48 Hs.528836 AA005023 NOD27 nucleotide-binding oligomerization domains 27 2.43 Hs.360940 AL589866 dJ222E13.1 kraken-like 2.40 Hs.515575 AI743780 PLAC2 placenta-specific 2 2.37 Hs.25674 AI827820 MBD2 methyl-CpG binding domain protein 2 2.36 Hs.12229 AA149594 TIEG2 TGFB inducible early growth response 2 2.33 Hs.482730 AA053711 EDIL3 EGF-like repeats and discoidin I-like domains 3 2.32 Hs. 370771 NM_000389 CDKN1A cyclin- depen dent kinase in hibitor 1A (p 21, Cip 1) 2282.28 Hs.469175 AI341537 JFC1 NADPH oxidase-related, C2 domain-containing protein 2.28 Hs.514821 AF043341 CCL5 chemokine (C-C motif) ligand 5 2.27 Hs.591292 NM_023915 GPR87 G protein-coupled receptor 87 2.26 Hs.500483 NM_001613 ACTA2 actin, alpha 2, smooth muscle, aorta 2.24 Hs.632824 NM_006729 DIAPH2 diaphanous homolog 2 (Drosophila) 2.22 Hs.369430 NM_000919 PAM peptidylglycine -

Yeast Genome Gazetteer P35-65
gazetteer Metabolism 35 tRNA modification mitochondrial transport amino-acid metabolism other tRNA-transcription activities vesicular transport (Golgi network, etc.) nitrogen and sulphur metabolism mRNA synthesis peroxisomal transport nucleotide metabolism mRNA processing (splicing) vacuolar transport phosphate metabolism mRNA processing (5’-end, 3’-end processing extracellular transport carbohydrate metabolism and mRNA degradation) cellular import lipid, fatty-acid and sterol metabolism other mRNA-transcription activities other intracellular-transport activities biosynthesis of vitamins, cofactors and RNA transport prosthetic groups other transcription activities Cellular organization and biogenesis 54 ionic homeostasis organization and biogenesis of cell wall and Protein synthesis 48 plasma membrane Energy 40 ribosomal proteins organization and biogenesis of glycolysis translation (initiation,elongation and cytoskeleton gluconeogenesis termination) organization and biogenesis of endoplasmic pentose-phosphate pathway translational control reticulum and Golgi tricarboxylic-acid pathway tRNA synthetases organization and biogenesis of chromosome respiration other protein-synthesis activities structure fermentation mitochondrial organization and biogenesis metabolism of energy reserves (glycogen Protein destination 49 peroxisomal organization and biogenesis and trehalose) protein folding and stabilization endosomal organization and biogenesis other energy-generation activities protein targeting, sorting and translocation vacuolar and lysosomal -

Congenital Disorders of Glycosylation from a Neurological Perspective
brain sciences Review Congenital Disorders of Glycosylation from a Neurological Perspective Justyna Paprocka 1,* , Aleksandra Jezela-Stanek 2 , Anna Tylki-Szyma´nska 3 and Stephanie Grunewald 4 1 Department of Pediatric Neurology, Faculty of Medical Science in Katowice, Medical University of Silesia, 40-752 Katowice, Poland 2 Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, 01-138 Warsaw, Poland; [email protected] 3 Department of Pediatrics, Nutrition and Metabolic Diseases, The Children’s Memorial Health Institute, W 04-730 Warsaw, Poland; [email protected] 4 NIHR Biomedical Research Center (BRC), Metabolic Unit, Great Ormond Street Hospital and Institute of Child Health, University College London, London SE1 9RT, UK; [email protected] * Correspondence: [email protected]; Tel.: +48-606-415-888 Abstract: Most plasma proteins, cell membrane proteins and other proteins are glycoproteins with sugar chains attached to the polypeptide-glycans. Glycosylation is the main element of the post- translational transformation of most human proteins. Since glycosylation processes are necessary for many different biological processes, patients present a diverse spectrum of phenotypes and severity of symptoms. The most frequently observed neurological symptoms in congenital disorders of glycosylation (CDG) are: epilepsy, intellectual disability, myopathies, neuropathies and stroke-like episodes. Epilepsy is seen in many CDG subtypes and particularly present in the case of mutations -

(Glycogen Storage Disease Type V) and Anesthesia a Case Report And
Pediatric Anesthesia ISSN 1155-5645 REVIEW ARTICLE McArdle’s disease (glycogen storage disease type V) and anesthesia – a case report and review of the literature Georg Bollig1,2 1 Department of Anesthesiology and Intensive Care, Palliative Medicine and Pain Therapy, HELIOS Klinikum Schleswig, Schleswig, Germany 2 Department of Surgical Sciences, Haukeland University Hospital, University of Bergen, Bergen, Norway Keywords Summary general anesthesia; glycogen storage disease; glycogen storage disease type V; McArdles disease (glycogen storage disease type v) is a rare condition in malignant hyperthermia; McArdles disease; which energy-metabolism in the muscle is hampered. A case report is pre- perioperative complications sented and the possible risk for perioperative complications including malig- nant hyperthermia is discussed. A checklist for the anesthesiological Correspondence management of patients with McArdles disease is provided. A short overview Georg Bollig, Department of Anesthesiology of anesthesiological challenges and perioperative complications of other gly- and Intensive Care, Palliative Medicine and Pain Therapy, HELIOS Klinikum Schleswig, cogen storage diseases is given. Schleswig, Germany Email: [email protected] Section Editor: Barbara Brandom Accepted 3 March 2013 doi:10.1111/pan.12164 tachycardia and hypotonia occurred. After the opera- Introduction tion, the anesthesiologist informed the patient about the McArdle’s disease is a rare condition in which energy event and the fact that he had elevated liver enzymes metabolism in the muscle is hampered. A case report (aspartate transaminase = AST, alanine transaminase = will be presented, and the possible risk of perioperative ALT, and lactate dehydrogenase = LDH). An overview problems including malignant hyperthermia is dis- of the patient’s anesthesiological history is given in cussed. -

NICU Gene List Generator.Xlsx
Neonatal Crisis Sequencing Panel Gene List Genes: A2ML1 - B3GLCT A2ML1 ADAMTS9 ALG1 ARHGEF15 AAAS ADAMTSL2 ALG11 ARHGEF9 AARS1 ADAR ALG12 ARID1A AARS2 ADARB1 ALG13 ARID1B ABAT ADCY6 ALG14 ARID2 ABCA12 ADD3 ALG2 ARL13B ABCA3 ADGRG1 ALG3 ARL6 ABCA4 ADGRV1 ALG6 ARMC9 ABCB11 ADK ALG8 ARPC1B ABCB4 ADNP ALG9 ARSA ABCC6 ADPRS ALK ARSL ABCC8 ADSL ALMS1 ARX ABCC9 AEBP1 ALOX12B ASAH1 ABCD1 AFF3 ALOXE3 ASCC1 ABCD3 AFF4 ALPK3 ASH1L ABCD4 AFG3L2 ALPL ASL ABHD5 AGA ALS2 ASNS ACAD8 AGK ALX3 ASPA ACAD9 AGL ALX4 ASPM ACADM AGPS AMELX ASS1 ACADS AGRN AMER1 ASXL1 ACADSB AGT AMH ASXL3 ACADVL AGTPBP1 AMHR2 ATAD1 ACAN AGTR1 AMN ATL1 ACAT1 AGXT AMPD2 ATM ACE AHCY AMT ATP1A1 ACO2 AHDC1 ANK1 ATP1A2 ACOX1 AHI1 ANK2 ATP1A3 ACP5 AIFM1 ANKH ATP2A1 ACSF3 AIMP1 ANKLE2 ATP5F1A ACTA1 AIMP2 ANKRD11 ATP5F1D ACTA2 AIRE ANKRD26 ATP5F1E ACTB AKAP9 ANTXR2 ATP6V0A2 ACTC1 AKR1D1 AP1S2 ATP6V1B1 ACTG1 AKT2 AP2S1 ATP7A ACTG2 AKT3 AP3B1 ATP8A2 ACTL6B ALAS2 AP3B2 ATP8B1 ACTN1 ALB AP4B1 ATPAF2 ACTN2 ALDH18A1 AP4M1 ATR ACTN4 ALDH1A3 AP4S1 ATRX ACVR1 ALDH3A2 APC AUH ACVRL1 ALDH4A1 APTX AVPR2 ACY1 ALDH5A1 AR B3GALNT2 ADA ALDH6A1 ARFGEF2 B3GALT6 ADAMTS13 ALDH7A1 ARG1 B3GAT3 ADAMTS2 ALDOB ARHGAP31 B3GLCT Updated: 03/15/2021; v.3.6 1 Neonatal Crisis Sequencing Panel Gene List Genes: B4GALT1 - COL11A2 B4GALT1 C1QBP CD3G CHKB B4GALT7 C3 CD40LG CHMP1A B4GAT1 CA2 CD59 CHRNA1 B9D1 CA5A CD70 CHRNB1 B9D2 CACNA1A CD96 CHRND BAAT CACNA1C CDAN1 CHRNE BBIP1 CACNA1D CDC42 CHRNG BBS1 CACNA1E CDH1 CHST14 BBS10 CACNA1F CDH2 CHST3 BBS12 CACNA1G CDK10 CHUK BBS2 CACNA2D2 CDK13 CILK1 BBS4 CACNB2 CDK5RAP2 -

Guaiacol As a Drug Candidate for Treating Adult Polyglucosan Body Disease
Guaiacol as a drug candidate for treating adult polyglucosan body disease Or Kakhlon, … , Wyatt W. Yue, H. Orhan Akman JCI Insight. 2018;3(17):e99694. https://doi.org/10.1172/jci.insight.99694. Research Article Metabolism Therapeutics Graphical abstract Find the latest version: https://jci.me/99694/pdf RESEARCH ARTICLE Guaiacol as a drug candidate for treating adult polyglucosan body disease Or Kakhlon,1 Igor Ferreira,2 Leonardo J. Solmesky,3 Netaly Khazanov,4 Alexander Lossos,1 Rafael Alvarez,5 Deniz Yetil,6 Sergey Pampou,7 Miguel Weil,3,8 Hanoch Senderowitz,4 Pablo Escriba,5 Wyatt W. Yue,2 and H. Orhan Akman9 1Department of Neurology, Hadassah-Hebrew University Medical Center, Jerusalem, Israel. 2Structural Genomics Consortium, Nuffield Department of Clinical Medicine, University of Oxford, Oxford, United Kingdom.3 Cell Screening Facility for Personalized Medicine, Department of Cell Research and Immunology, The George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv, Israel. 4Department of Chemistry, Bar Ilan University, Ramat Gan, Israel. 5Laboratory of Molecular Cell Biomedicine, Department of Biology, University of the Balearic Islands, Palma de Mallorca, Spain. 6Connecticut College, Newington, Connecticut USA. 7Columbia University Department of Systems Biology Irving Cancer Research Center, New York, New York, USA. 8Laboratory for Neurodegenerative Diseases and Personalized Medicine, Department of Cell Research and Immunology, The George S. Wise Faculty for Life Sciences, Sagol School of Neurosciences, Tel Aviv University, Ramat Aviv, Tel Aviv, Israel. 9Columbia University Medical Center Department of Neurology, Houston Merritt Neuromuscular diseases research center, New York, New York, USA. Adult polyglucosan body disease (APBD) is a late-onset disease caused by intracellular accumulation of polyglucosan bodies, formed due to glycogen-branching enzyme (GBE) deficiency. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Supplementary Table 1. in Vitro Side Effect Profiling Study for LDN/OSU-0212320. Neurotransmitter Related Steroids
Supplementary Table 1. In vitro side effect profiling study for LDN/OSU-0212320. Percent Inhibition Receptor 10 µM Neurotransmitter Related Adenosine, Non-selective 7.29% Adrenergic, Alpha 1, Non-selective 24.98% Adrenergic, Alpha 2, Non-selective 27.18% Adrenergic, Beta, Non-selective -20.94% Dopamine Transporter 8.69% Dopamine, D1 (h) 8.48% Dopamine, D2s (h) 4.06% GABA A, Agonist Site -16.15% GABA A, BDZ, alpha 1 site 12.73% GABA-B 13.60% Glutamate, AMPA Site (Ionotropic) 12.06% Glutamate, Kainate Site (Ionotropic) -1.03% Glutamate, NMDA Agonist Site (Ionotropic) 0.12% Glutamate, NMDA, Glycine (Stry-insens Site) 9.84% (Ionotropic) Glycine, Strychnine-sensitive 0.99% Histamine, H1 -5.54% Histamine, H2 16.54% Histamine, H3 4.80% Melatonin, Non-selective -5.54% Muscarinic, M1 (hr) -1.88% Muscarinic, M2 (h) 0.82% Muscarinic, Non-selective, Central 29.04% Muscarinic, Non-selective, Peripheral 0.29% Nicotinic, Neuronal (-BnTx insensitive) 7.85% Norepinephrine Transporter 2.87% Opioid, Non-selective -0.09% Opioid, Orphanin, ORL1 (h) 11.55% Serotonin Transporter -3.02% Serotonin, Non-selective 26.33% Sigma, Non-Selective 10.19% Steroids Estrogen 11.16% 1 Percent Inhibition Receptor 10 µM Testosterone (cytosolic) (h) 12.50% Ion Channels Calcium Channel, Type L (Dihydropyridine Site) 43.18% Calcium Channel, Type N 4.15% Potassium Channel, ATP-Sensitive -4.05% Potassium Channel, Ca2+ Act., VI 17.80% Potassium Channel, I(Kr) (hERG) (h) -6.44% Sodium, Site 2 -0.39% Second Messengers Nitric Oxide, NOS (Neuronal-Binding) -17.09% Prostaglandins Leukotriene,