Advances in the Asymmetric Total Synthesis of Natural Products Using Chiral Secondary Amine Catalyzed Reactions of Α,Β-Unsaturated Aldehydes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
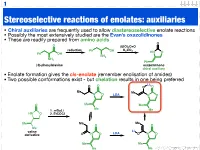
Stereoselective Reactions of Enolates: Auxiliaries
1 Stereoselective reactions of enolates: auxiliaries • Chiral auxiliaries are frequently used to allow diastereoselective enolate reactions • Possibly the most extensively studied are the Evan’s oxazolidinones • These are readily prepared from amino acids O O (EtO)2C=O reduction K CO Ph OH 2 3 HN O Ph OH NH2 NH2 Ph (S)-phenylalanine oxazolidinone chiral auxiliary • Enolate formation gives the cis-enolate (remember enolisation of amides) • Two possible conformations exist - but chelation results in one being preferred Li O O O O Me Me N O LDA N O O Me Me Me 1. n-BuLi Me HN O 2. EtCOCl Me Me Me O O Me Li valine LDA derivative O N O O N O Me Me Me Me 123.702 Organic Chemistry 2 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 3 Diastereoselective alkylation of Evan’s enolate Li O O O O Me Me N O PhCH2I N O Ph Me Me Me Me I Ph Li O Bn O O O O O H H Me N Me N H Me Me Me Me iso-propyl group blocks bottom face • Clearly (I hope) one face of the enolate is blocked • Chelation results in a rigid structure that provides maximum steric hindrance • The electrophile can only approach from one face 123.702 Organic Chemistry 4 Diastereoselective functionalisation Li O O O O Br LDA Me Me N O N O H Ph Ph 96% de H O -

Synthesis of FMN Analogues As Probes for the Photocycle of Avena Sativa
Synthesis of FMN Analogues as Probes for the Photocycle of Avena sativa Andrew Wood Submitted in fulfilment of the requirement for the degree of Doctor of Philosophy School of Chemistry, Cardiff University February, 2014 Declaration This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 1 This thesis is being submitted in partial fulfilment of the requirements for the degree of PhD. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated (see below). Other sources are acknowledged by explicit references. The views expressed are my own. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter- library loan, and for the title and summary to be made available to outside organisations. Signed ………………………………………… (candidate) Date ………………………… i Declaration of Contributions I confirm that the work presented within this thesis was performed by the author, with exceptions summarised here, and explicitly referenced in the relevant sections of the thesis. The original construction of several plasmid vectors used for the expression of recombinant proteins was not performed, as they were freely available (from previous work), and donated to me. Their original preparation is summarised below. Furthermore, during a short side- project the dephosphorylation of 5-deazaFAD was performed (on an analytical scale) by a collaborator (Dr. Hannah Collins, University of Kent) using commercially available Phosphodiesterase I from Crotalus atrox. -

Brittain-DR-1965-Phd-Thesis.Pdf
POLYMETHYLENE PYRIDINES , A thesis submitted by David Robert Brittain in partial fulfilment of the requirements for the degree of DOCTOR OP PHILOSOPHY in the University of London Organic Chemistry Department, dune, 1965. Imperial College, LONDON, S.W.7. ABSTRACT This thesis describes a series of attempts to syn- thesise 2,5- and 1,4-polymethylene bridged pyridines. Nuclear magnetic resonance theory predicts that protons, which are held directly over an aromatic ring, will be abnormally shielded compared with protons in aliphatic straight-chain hydrocarbons. This prediction has been verified for the central methylene protons of paracyclo- phanes. The degree of shielding, expressed in terms of the distance from the aromatic ring, is a measure of the induced ring current and hence the aromaticity of the benzene ring. Similar measurements upon 2,5- or 1,4— polymethylene bridged pyridines would make it possible to determine the degree of aromaticity of the pyridine ring relative to benzene. A review of the subject of aromaticity is presented in which special reference has been made to its inter- pretation by nuclear magnetic resonance. The synthetic work has not been brougL.t to a truly satisfactory conclusion. However, the synthetic routes to 2,5-dialkylpyridines have been thoroughly investigated and a wide variety of such compounds prepared. The functional groups at the ends of the alkyl chains have been varied in an effort to produce a derivative which would cyclise to give a 2,5-bridged pyridine. The attempted intramolecular oxidative coupling of 2,5-dihex- 51 -ynylpyridine received much attention. In the attempts to obtain a 1,4-bridged pyridine, two tricyclic compounds, each containing two quaternised pyridine rings linked by polymethylene chains, were obtained. -

Catalytic Direct Asymmetric Michael Reactions
ORGANIC LETTERS 2001 Catalytic Direct Asymmetric Michael Vol. 3, No. 23 Reactions: Taming Naked Aldehyde 3737-3740 Donors Juan M. Betancort and Carlos F. Barbas III* The Skaggs Institute for Chemical Biology and the Department of Molecular Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037 [email protected] Received September 5, 2001 ABSTRACT Direct catalytic enantio- and diastereoselective Michael addition reactions of unmodified aldehydes to nitro olefins using (S)-2-(morpholinomethyl)- pyrrolidine as a catalyst are described. The reactions proceed in good yield (up to 96%) in a highly syn-selective manner (up to 98:2) with enantioselectivities approaching 80%. The resulting γ-formyl nitro compounds are readily converted to chiral, nonracemic 3,4-disubstituted pyrrolidines. The Michael reaction is generally regarded as one of the Typically, carbon nucleophiles that contain an active most efficient carbon-carbon bond forming reactions, and methylene center such as malonic acid esters, â-keto esters, studies concerning this reaction have played an important nitroalkanes, etc. have been studied in the Michael reaction. role in the development of modern synthetic organic Carbonyl compounds, and ketones in particular, have gener- chemistry.1 As the demand for optically active compounds ally only been used as donors following their preactivation has soared in recent years, much progress has been made in by conversion into a more reactive species such as enol or the development of asymmetric variants of this reaction, enamine equivalents.5,6 In these cases, additional synthetic providing for the preparation of Michael adducts with high enantiomeric purity.2 Though remarkable advances have been (3) (a) Chataigner, I.; Gennari, C.; Ongeri, S.; Piarulli, U.; Ceccarelli, S. -

Electrooxidation Enables Highly Regioselective Dearomative Annulation of Indole and Benzofuran Derivatives
ARTICLE https://doi.org/10.1038/s41467-019-13829-4 OPEN Electrooxidation enables highly regioselective dearomative annulation of indole and benzofuran derivatives Kun Liu1, Wenxu Song1, Yuqi Deng1, Huiyue Yang1, Chunlan Song1, Takfaoui Abdelilah1, Shengchun Wang 1, Hengjiang Cong 1, Shan Tang1 & Aiwen Lei1* 1234567890():,; The dearomatization of arenes represents a powerful synthetic methodology to provide three-dimensional chemicals of high added value. Here we report a general and practical protocol for regioselective dearomative annulation of indole and benzofuran derivatives in an electrochemical way. Under undivided electrolytic conditions, a series of highly functio- nalized five to eight-membered heterocycle-2,3-fused indolines and dihydrobenzofurans, which are typically unattainable under thermal conditions, can be successfully accessed in high yield with excellent regio- and stereo-selectivity. This transformation can also tolerate a wide range of functional groups and achieve good efficiency in large-scale synthesis under oxidant-free conditions. In addition, cyclic voltammetry, electron paramagnetic resonance (EPR) and kinetic studies indicate that the dehydrogenative dearomatization annulations arise from the anodic oxidation of indole into indole radical cation, and this process is the rate- determining step. 1 College of Chemistry and Molecular Sciences, Institute for Advanced Studies (IAS), Wuhan University, Wuhan 430072, P. R. China. *email: aiwenlei@whu. edu.cn NATURE COMMUNICATIONS | (2020) 11:3 | https://doi.org/10.1038/s41467-019-13829-4 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-019-13829-4 reaking the aromatic systems of electron-rich arenes or fused indolines (Fig. 1a)39–44. Therefore, it is highly appealing to heteroarenes provides three-dimensional chemicals of high develop efficient approaches to allow for their preparation. -

A General and Direct Reductive Amination of Aldehydes and Ketones with Electron-Deficient Anilines
SYNTHESIS0039-78811437-210X © Georg Thieme Verlag Stuttgart · New York 2016, 48, 1301–1317 1301 feature Syn thesis J. Pletz et al. Feature A General and Direct Reductive Amination of Aldehydes and Ketones with Electron-Deficient Anilines Jakob Pletz EWG hydride reductant EWG H NH 1 N R1 Bernhard Berg 2 O R activating agent + Rolf Breinbauer* 2 R2 R Institute of Organic Chemistry, Graz University of Technology, 29 examples up to 99% yield Stremayrgasse 9, 8010 Graz, Austria 3 methods: [email protected] A) BH3•THF, AcOH, CH2Cl2, 0–20 °C 12 anilines In memoriam Philipp Köck B) BH3•THF, TMSCl, DMF, 0 °C 14 ketones C) NaBH4, TMSCl, DMF, 0 °C 3 aldehydes Received: 01.12.2015 Our first goal was the design and synthesis of analogues Accepted after revision: 20.01.2016 of intermediates in the transformation catalyzed by the Published online: 01.03.2016 1 DOI: 10.1055/s-0035-1561384; Art ID: ss-2015-t0688-fa protein PhzA/B. According to Wulf’s proposal, this enzyme was expected to catalyze the imine formation between the putative aminoketone B, resulting from the transformation Abstract In our ongoing efforts in preparing tool compounds for in- of PhzF with dihydrohydroxyanthranilic acid (DHHA; A), vestigating and controlling the biosynthesis of phenazines, we recog- with itself, thereby establishing the tricyclic skeleton C, nized the limitations of existing protocols for C–N bond formation of from which after a series of oxidation reactions phenazine- electron-deficient anilines when using reductive amination. After ex- carboxylic acid E will -

Page 1 of 108 RSC Advances
RSC Advances This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. This Accepted Manuscript will be replaced by the edited, formatted and paginated article as soon as this is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/advances Page 1 of 108 RSC Advances Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis Majid M. Heravi,* Vahideh Zadsirjan, Behnaz Farajpour Department of Chemistry, School of Science, Alzahra University, Vanak, Tehran, Iran Email: [email protected] Abstract Various chiral oxazolidinones (Evans' oxazolidinones) have been employed as effective chiral auxiliaries in the asymmetric alkylation of different enolates. This strategy has been found promising and successful when used as key step (steps) in the total synthesis of several biologically active natural products. In this report, we try to underscore the applications of Manuscript oxazolidinones as chiral auxiliary in asymmetric alkylation, and particularly in crucial chiral inducing steps in the total synthesis of natural products, showing biological activities. -

Aldol Reactions: E-Enolates and Anti-Selectivity
Utah State University DigitalCommons@USU All Graduate Plan B and other Reports Graduate Studies 5-2005 Aldol Reactions: E-Enolates and Anti-Selectivity Matthew Grant Anderson Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/gradreports Part of the Organic Chemistry Commons Recommended Citation Anderson, Matthew Grant, "Aldol Reactions: E-Enolates and Anti-Selectivity" (2005). All Graduate Plan B and other Reports. 1312. https://digitalcommons.usu.edu/gradreports/1312 This Report is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Plan B and other Reports by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. ALDOL REACTIONS: E-ENOLATES AND ANTI-SELECTIVITY Prepared By: MATTHEW GRANT ANDERSON A non-thesis paper submitted in partial fulfillment of the requirement for a Plan B Degree of Masters of Science in Organic Chemistry UTAH STATE UNIVERSITY Logan, Utah 2005 Contents Page CONTENTS ...................................................................................... .i LIST OF TABLES, FIGURES AND SCHEMES ....................................... ii,iii ABSTRACT .................................................................................... iv CHAPTER I. ALDOL REACTIONS:E-ENOLATES AND ANTI SELECTIVITY ......... 1 CHAPTER II. SECTION 1. MODELS OF E-ENOLATE FORMATION ...... .... ....... ... 12 SECTION 2. PATERSON ENOLATE PAPER ..... ......................... -

Robert Burns Woodward
The Life and Achievements of Robert Burns Woodward Long Literature Seminar July 13, 2009 Erika A. Crane “The structure known, but not yet accessible by synthesis, is to the chemist what the unclimbed mountain, the uncharted sea, the untilled field, the unreached planet, are to other men. The achievement of the objective in itself cannot but thrill all chemists, who even before they know the details of the journey can apprehend from their own experience the joys and elations, the disappointments and false hopes, the obstacles overcome, the frustrations subdued, which they experienced who traversed a road to the goal. The unique challenge which chemical synthesis provides for the creative imagination and the skilled hand ensures that it will endure as long as men write books, paint pictures, and fashion things which are beautiful, or practical, or both.” “Art and Science in the Synthesis of Organic Compounds: Retrospect and Prospect,” in Pointers and Pathways in Research (Bombay:CIBA of India, 1963). Robert Burns Woodward • Graduated from MIT with his Ph.D. in chemistry at the age of 20 Woodward taught by example and captivated • A tenured professor at Harvard by the age of 29 the young... “Woodward largely taught principles and values. He showed us by • Published 196 papers before his death at age example and precept that if anything is worth 62 doing, it should be done intelligently, intensely • Received 24 honorary degrees and passionately.” • Received 26 medals & awards including the -Daniel Kemp National Medal of Science in 1964, the Nobel Prize in 1965, and he was one of the first recipients of the Arthur C. -

Recommended Methods for the Identification and Analysis Of
Vienna International Centre, P.O. Box 500, 1400 Vienna, Austria Tel: (+43-1) 26060-0, Fax: (+43-1) 26060-5866, www.unodc.org RECOMMENDED METHODS FOR THE IDENTIFICATION AND ANALYSIS OF AMPHETAMINE, METHAMPHETAMINE AND THEIR RING-SUBSTITUTED ANALOGUES IN SEIZED MATERIALS (revised and updated) MANUAL FOR USE BY NATIONAL DRUG TESTING LABORATORIES Laboratory and Scientific Section United Nations Office on Drugs and Crime Vienna RECOMMENDED METHODS FOR THE IDENTIFICATION AND ANALYSIS OF AMPHETAMINE, METHAMPHETAMINE AND THEIR RING-SUBSTITUTED ANALOGUES IN SEIZED MATERIALS (revised and updated) MANUAL FOR USE BY NATIONAL DRUG TESTING LABORATORIES UNITED NATIONS New York, 2006 Note Mention of company names and commercial products does not imply the endorse- ment of the United Nations. This publication has not been formally edited. ST/NAR/34 UNITED NATIONS PUBLICATION Sales No. E.06.XI.1 ISBN 92-1-148208-9 Acknowledgements UNODC’s Laboratory and Scientific Section wishes to express its thanks to the experts who participated in the Consultative Meeting on “The Review of Methods for the Identification and Analysis of Amphetamine-type Stimulants (ATS) and Their Ring-substituted Analogues in Seized Material” for their contribution to the contents of this manual. Ms. Rosa Alis Rodríguez, Laboratorio de Drogas y Sanidad de Baleares, Palma de Mallorca, Spain Dr. Hans Bergkvist, SKL—National Laboratory of Forensic Science, Linköping, Sweden Ms. Warank Boonchuay, Division of Narcotics Analysis, Department of Medical Sciences, Ministry of Public Health, Nonthaburi, Thailand Dr. Rainer Dahlenburg, Bundeskriminalamt/KT34, Wiesbaden, Germany Mr. Adrian V. Kemmenoe, The Forensic Science Service, Birmingham Laboratory, Birmingham, United Kingdom Dr. Tohru Kishi, National Research Institute of Police Science, Chiba, Japan Dr. -

Aldol Condensation
Chemistry 212 Laboratory Dibenzalacetone via Crossed Aldol Condensation Prelab: Calculate the amounts of all chemicals needed in measurable amounts (i.e. grams or milliliters rather than moles.) Introduction: Aldol condensations are important in organic synthesis, providing a good way to form carbon–carbon bonds. The "aldol" (aldehyde + alcohol) product is a structural unit found in many naturally occurring molecules and pharmaceuticals, and is therefore important. In an Aldol condensation an enolate ion reacts with a carbonyl compound to form a β- hydroxyaldehyde or β-hydroxyketone, followed by dehydration to give a conjugated enone. The general equation is shown in Figure 1. O O O R" B: H R R'" R "R R'" loss of H2O H R' R' Figure 1. The equation for the Aldol Condensation. The reaction involves the nucleophilic addition of an enolate to an aldehyde to form a β-hydroxy carbonyl. The β-hydroxy carbonyl is readily dehydrated under mild conditions. The aldol reaction occurs under both acidic and basic conditions as seen in Figure 2. ENOL pathway (reacts in H O protonated OH form) O O catalytic H+ O O H H R' H2O lost R' R R R' R R H aldol addition product aldol condensation product ENOLATE pathway O O M O M O base O H R' R R' R R enolate H Figure 2. The Aldol reaction and subsequent dehydration under acidic and basic conditions. The reaction we will be doing this week involves the reaction between benzaldehyde and acetone to do a double Aldol Condensation. The overall equation is shown in Figure 3. -

Annulation of Propargylic Dithioacetals Leading to Furan-Containing Oligoaryls*
Pure Appl. Chem., Vol. 77, No. 7, pp. 1213–1219, 2005. DOI: 10.1351/pac200577071213 © 2005 IUPAC Annulation of propargylic dithioacetals leading to furan-containing oligoaryls* Tien-Yau Luh Department of Chemistry, National Taiwan University, Taipei 106, Taiwan Abstract: Reaction of propargylic dithioacetals with BuLi followed by treatment with di- aldehydes yields the corresponding allenyl carbinols which can be cyclized to give the 2,3,5-trisubstituted furans. The use of this strategy for the synthesis of a range of alternating benzene-furan oligoaryls is described. Keywords: annulation; propargylic dithioacetals; oligoaryls; furans; optoelectronic applica- tions; cyclophanes; convergent synthesis. INTRODUCTION Direct conversion of carbon–sulfur bonds in dithioacetal functionality into the corresponding car- bon–carbon bonds has been extensively studied in recent years [1]. Representative examples are shown in Scheme 1. The substituents R1 and/or R2 can be aryl, alkenyl alkynyl, or, more recently, alkyl groups. Since carbon and sulfur atoms have similar electronegativities, the polarity of the carbon–sulfur bond can vary depending on the nature of the substrates and reaction conditions. In general, the carbon end of a carbon–sulfur bond can serve as a carbocationic equivalent to reaction with a nucleophile. Alternatively, the nucleophile can attack at the sulfur end of a carbon–sulfur bond to generate the car- banion, which can then react with an electrophile leading to the corresponding substitution product. In this case, the carbanionic moiety must be stabilized by a neighboring stabilizing substituent(s). Ikehira and Krief have shown that benzylic dithiolanes react with butyllithium followed by protonolysis to give the corresponding mono-desulfurized products (eq.