Brittain-DR-1965-Phd-Thesis.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

1St Year Students Lecture No. 5 Ahmed Mohamad Jawad
1st year students Lecture No. 5 Ahmed Mohamad Jawad Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Out line 1- Dienes: Introduction of diene Nomenclature of diene Preparation of diene Reaction of diene 2- Alkyne Introduction of Alkyne Nomenclature of Alkyne Physical properties of alkyne Preparation of Alkyne Reaction of Alkyne Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene Dienes are simply alkenes that contain two carbon-carbon double bonds. Dienes are divided into two major important classes according to the arrangement of the double bonds 1-Conjugated : Double bonds that alternate with single bonds are said to be conjugated. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene 2-Isolated double bonds that are separated by more than one single bond are said to be isolated Less stable than conjugated 3-Cumulated : contains cumulated double bonds Least one stable . Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Nomenclature of diene Dienes are named by the IUPAC system in the same way as alkenes , except that the ending diene is used, with two numbers to indicate the position of the two double bonds. H2C C CH2 1,2-propadiene 1,3-butadiene 1,4-pentadiene This system is easily extended to compounds containing any number of double bonds. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Preparation of diene 1- by acid catalyzed double dehydration 2- By dehydrogenation of dihalides: Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Reactions of Dienes In general terms, dienes undergo electrophilic addition reactions in a similar approach of alkenes Conjugated dienes undergo addition but the proximity of the conjugated C=C influences the reactions. -

Photoremovable Protecting Groups Used for the Caging of Biomolecules
1 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1 2-Nitrobenzyl and 7-Nitroindoline Derivatives John E.T. Corrie 1.1.1 Introduction 1.1.1.1 Preamble and Scope of the Review This chapter covers developments with 2-nitrobenzyl (and substituted variants) and 7-nitroindoline caging groups over the decade from 1993, when the author last reviewed the topic [1]. Other reviews covered parts of the field at a similar date [2, 3], and more recent coverage is also available [4–6]. This chapter is not an exhaustive review of every instance of the subject cages, and its principal focus is on the chemistry of synthesis and photocleavage. Applications of indi- vidual compounds are only briefly discussed, usually when needed to put the work into context. The balance between the two cage types is heavily slanted to- ward the 2-nitrobenzyls, since work with 7-nitroindoline cages dates essentially from 1999 (see Section 1.1.3.2), while the 2-nitrobenzyl type has been in use for 25 years, from the introduction of caged ATP 1 (Scheme 1.1.1) by Kaplan and co-workers [7] in 1978. Scheme 1.1.1 Overall photolysis reaction of NPE-caged ATP 1. Dynamic Studies in Biology. Edited by M. Goeldner, R. Givens Copyright © 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30783-4 2 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1.1.2 Historical Perspective The pioneering work of Kaplan et al. [7], although preceded by other examples of 2-nitrobenzyl photolysis in synthetic organic chemistry, was the first to apply this to a biological problem, the erythrocytic Na:K ion pump. -

Diels-Alder Reaction Between Naphthalene and N-Phenylmaleimide Under Ambient and High Pressure Conditions
Issue in Honor of Prof. Alexander I. Konovalov ARKIVOC 2004 (xii) 70-79 Diels-Alder reaction between naphthalene and N-phenylmaleimide under ambient and high pressure conditions Gulnara G. Iskhakova*, Vladimir D. Kiselev, Elena A. Kashaeva, Lubov’ N. Potapova, Evgeny A. Berdnikov, Dmitry B. Krivolapov, and Igor A. Litvinov A. M. Butlerov Chemical Institute, Kazan State University, Kremlevskaya str.18, 420008 Kazan, Russian Federation E-mail: [email protected] Dedicated to the 70th anniversary of Professor Alexander I. Konovalov (received 02 Nov 04; accepted 08 Jan 05; published on the web 05 Feb 05) Abstract The rate and equilibrium constants for the Diels-Alder reactions between benzene and naphthalene as dienes and tetracyanoethylene, maleic anhydride and N-phenylmaleimide as dienophiles at 25 °С were estimated from empirical rule. The highest yield of the adduct was predicted for the reaction of naphthalene with N-phenylmaleimide. The time of adduct formation in 50% yield exceeds 30 years. The use of gallium chloride as a catalyst affords the exo-adduct for seven days at room temperature. The rate ((2±0.5)•10-6 L mol-1 s-1) and equilibrium constants (5±2 L mol-1) of this reaction were determined. Under high pressure conditions (8 kbar) reaction occurs with formation of both stereo isomers at 100 °С during 80 hours. Keywords: Naphthalene, Diels-Alder reaction, catalysis, high pressure Introduction 1 Many dienes, including substituted benzenes and naphthalenes, are known to form molecular complexes of the π,π-type due to the interaction of the highest occupied π-orbital of a diene- donor with the lowest unoccupied π-orbital of a dienophile-acceptor. -
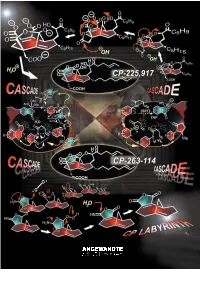
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

JUN 1 51998 Science
Efforts Toward the Syntheses of Natural Products: Part A: Paeoniflorin Part B: (+)-Taxusin A thesis presented by Rebecca J. Carazza B.S. Chemistry University of Massachusetts, Amherst, 1993 Submitted to the Department of Chemistry in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry at the Massachusetts Institute of Technology June 1998 © 1998 Massachusetts Institute of Technology. All rights reserved. SinatureofAuthor: SignatureT- -~- -- .. -uof -Author: -- I- ----- D4pdrtment o('ihemistry May 26, 1998 Certified by: ,Scott C. Virgil Thesis Advisor Acceuted bv: Dietmar Seyferth Chairman, Departmental Committee on Graduate Students O-V .Z\ . JUN 1 51998 Science UR PAES This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor Rick L. Danheiser - Chairman Professor Scott C. Virgil / Th/sis Supervisor Professor Peter H. Seeberger Efforts Toward the Syntheses of Natural Products: Part A: Paeoniflorin Part B: (+)-Taxusin by Rebecca J. Carazza Submitted to the Department of Chemistry on May 26, 1998 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemistry Massachusetts Institute of Technology ABSTRACT Part A Efforts toward the synthesis of the monoterpene glycoside paeoniflorin (1) are discussed. Optimization of the previous synthetic route was successful. Synthesis of the key cc-diazo intermediate 30 was achieved and the construction of the carbocyclic frame was completed. Key reactions of the strategy involve a ring contraction via a Wolff rearrangement, and formation of the lactone 78 which undergoes diisobutylaluminum hydride reduction followed by acid catalyzed cyclization to the paeoniflorin ring system 80. A new synthetic strategy was initiated to prepare the key [3.2.1]bicyclooctanone 81 using a palladium mediated olefin cyclization of the acyloin substrate 82. -

Industrial Chemistry Process Scheme for Sections 3 - 3.3.2 Butenes Natural Gas Lwet I Oil
Olefins - Part 1 Industrial Chemistry Process Scheme for Sections 3 - 3.3.2 Butenes Natural Gas lwet I Oil Gas Gas Naphtha Oil Vacuum Distillation iiHydrotreating Naphtha Catalytic and Steam Thermal Crack- Process &--Crac kin ------l Where they come from Ethylenenm Propene Acetylene Ethylene Fuel Gas Ethylene Propene CL- C5- Pyrolysis Fraction Fraction Gasoline , + I 112.2.1 CL-Riffinate C5- Raffinate Ethylene 2-Butene manufacture Ratfinate I1 isobutene or n- Butenes n- and lsopentenes n-Pentenes lsobutene Oligomers n- and lsobutane n- and lsopentanes n- and lsopentanes 68 3. Olejins most valuable C4 olefin source: As shown in Table 3-3, a significantly higher fraction of steam cracking of naphtha butenes is obtained from steam cracking of naphtha than from Which C4 are obtainedcatalytic by cracking steam of gas oil. Therefore cracking? naphtha steam cracking is the more interesting technology for production of unsatu- rated C4 compounds. decrease of total C4 and C4 olefins, but As the cracking- severity increases, both the total yield of the increase Of butadiene under high severity C4 fraction and the proportion of butenes decrease, while the cracking conditions (consequence of butanedesired higher C2H4isobutane yield) proportion of butadiene increases due to its higher stability: Table 3-3. Composition of Cq fractions from steam cracking of naphtha and catalytic cracking of gas oil (in wt%). but-1-ene (E)-but-2-ene Steam cracking Catalytic cracking 1-butene Cracked products Low High (FCC) zeolite trans-2-butene severity seventy catalyst -

Organic Seminar Abstracts
L I B RA R.Y OF THE UN IVE.R.SITY Of ILLI NOIS 547 l£6s \ 954/55 PV Return this book on or before the Latest Date stamped below. University of Illinois Library Llf.1—H41 Digitized by the Internet Archive in 2012 with funding from University of Illinois Urbana-Champaign http://archive.org/details/organicsemi195455univ \7^ — SEMINAR TOPICS CHEMISTRY 435 I Semester 1954-55 The Quinolizinium Ion and Some of its Derivatives Harvey M. Loux, September 24 1 The Stereochemistry of Atropine and Cocaine Ho E . Knipmeyer, September 24 4 Recent Developments in the Chemistry of Cinnoline Derivatives Roger H. Kottke , October 1 8 Interpretation of Electrophiiic Aromatic Substitution and Solvolysis of Allylic and Benzhydryl Chlorides in Terms of Hyperconjugation Robert D. Stolow, October 1 11 The Decahydronaphtholic Acids and Their Relationship to the Decalols and the Decalylamines Robert J. Harder, October 8 14 Bis-Cyclopentadienyl Metal Compounds Edwin L. DeYoung, October 8 17 Ion-Pairs as Intermediates in Solvolysis Reactions Arthur H. Goldkamp, October 15 21 Synthesis of Morphine Mohan D. Nair, October 15 24 Structural and Geometrical Isomerism in the Oxidation of Azo Compounds D. F. Morrow, October 22 27 Stereochemical Aspects of Thermochromism John W. Johnson, Jr., October 22 30 A New Method for the Preparation of Olefins --The Pyrolysis of Sulfites F. M. Scheidt, October 29 33 Recent Studies of Macrocyclic Ring Systems: Cyclophanes Fred P. Hauck, Jr., October 29 36 Mechanism of the Para-Claisen Rearrangement Hugh H . Gibbs , November 5 40 A Proposed Mechanism for Basic c is -Dehydrohalogenat ion Robert M. -

II. Nomenclature Rules for Alkenes 1. the Parent Name Will Be the Longest
1 Lecture 9 II. Nomenclature Rules For Alkenes 1. The parent name will be the longest carbon chain that contains both carbons of the double bond. Drop the -ane suffix of the alkane name and add the –ene suffix. Never name the double bond as a prefix. If a double bond is present, you have an alkene, not an alkane. alkane + -ene = alkene 2. Begin numbering the chain at the end nearest the double bond. Always number through the double bond and identify its position in the longest chain with the lower number. In the older IUPAC rules the number for the double bond was placed in front of the stem name with a hyphen. Under the newer rules, the number for the double bond is placed right in front of “ene”, with hyphens. We will use the newer rules for specifying the location of pi bonds. 1 2 3456 H3CCHCH CH 2 CH2 CH3 hex-2-ene (newer rules) 2-hexene (older rules) 3. Indicate the position of any substituent group by the number of the carbon atom in the parent (longest) chain to which it is attached. CH 1 2 345 3 H3CCHCHCHCH2 CH CH3 6 7 CH3 Numbering is determined by the double bond, not the branches, because the double bond has 5,6-dimethylhept-3-ene (newer rules) higher priority than any alkyl branch. 5,6-dimethyl-3-heptene (older rules) 4. Number cycloalkenes so that the double bond is 1,2 (number through the double bond). Number in the direction about the ring so that the lowest number is used at the first point of difference. -

Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy Petr Klan,́*,†,‡ Tomaś̌solomek,̌ †,‡ Christian G
Review pubs.acs.org/CR Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy Petr Klan,́*,†,‡ Tomaś̌Solomek,̌ †,‡ Christian G. Bochet,§ Aurelień Blanc,∥ Richard Givens,⊥ Marina Rubina,⊥ Vladimir Popik,# Alexey Kostikov,# and Jakob Wirz∇ † Department of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic ‡ Research Centre for Toxic Compounds in the Environment, Faculty of Science, Masaryk University, Kamenice 3, 625 00 Brno, Czech Republic § Department of Chemistry, University of Fribourg, Chemin du Museé 9, CH-1700 Fribourg, Switzerland ∥ Institut de Chimie, University of Strasbourg, 4 rue Blaise Pascal, 67000 Strasbourg, France ⊥ Department of Chemistry, University of Kansas, 1251 Wescoe Hall Drive, 5010 Malott Hall, Lawrence, Kansas 66045, United States # Department of Chemistry, University of Georgia, Athens, Georgia 30602, United States ∇ Department of Chemistry, University of Basel, Klingelbergstrasse 80, CH-4056 Basel, Switzerland 7.3. Arylsulfonyl Group 160 7.4. Ketones: 1,5- and 1,6-Hydrogen Abstraction 160 7.5. Carbanion-Mediated Groups 160 7.6. Sisyl and Other Silicon-Based Groups 161 7.7. 2-Hydroxycinnamyl Groups 161 7.8. α-Keto Amides, α,β-Unsaturated Anilides, CONTENTS and Methyl(phenyl)thiocarbamic Acid 162 7.9. Thiochromone S,S-Dioxide 162 1. Introduction 119 7.10. 2-Pyrrolidino-1,4-Benzoquinone Group 162 2. Arylcarbonylmethyl Groups 121 7.11. Triazine and Arylmethyleneimino Groups 162 2.1. Phenacyl and Other Related Arylcarbonyl- 7.12. Xanthene and Pyronin Groups 163 methyl Groups 121 o 7.13. Retro-Cycloaddition Reactions 163 2.2. -Alkylphenacyl Groups 123 8. Sensitized Release 163 2.3. p-Hydroxyphenacyl Groups 125 p 8.1. -

Syntheses of Morphine and Codeine (1992 – 2002): Templates for Exploration of Synthetic Tools
Current Organic Synthesis, 2006, 3, 99-120 99 Syntheses of Morphine and Codeine (1992 – 2002): Templates for Exploration of Synthetic Tools L. M. Mascavage#, M. L. Wilson and D. R. Dalton* Department of Chemistry (016-00), Beury Hall, 13th and Norris Streets, Temple University, Philadelphia, PA 19122, USA and Department of Chemistry, Arcadia University, Glenside, PA, 19038, USA Abstract: Morphine (1) and its O-methylated analogue codeine (2), analgesic alkaloids of the opium poppy (Papaver Somniferium), have been targets of organic chemists engaged in synthetic activities for at least half a century. The “first” (Gates) and “most efficient” (Rice) syntheses of morphine (1) and codeine (2) are well known and have been reviewed and analyzed extensively numerous times. However, syntheses of the same two alkaloids that have been reported since 1992 and which have been used as devices to advance the art of organic synthesis are not as widely recognized and they have not been as thoroughly reviewed. Here they are analyzed in the spirit of the use of these two compounds as templates. Further, since both racemic and enantiospecific syntheses are important and since all eight (8) approaches (since 1992) are sufficiently different so as to warrant more tha n superficial examination, they are all considered. H HO 7 H 8 15 6 H H 6 14 N 5 CH3 H 13 NCH3 HO 14 13 9 16 9 O 16 12 15 10 O 10 4 4 11 1, R = H 2, R = CH 1 1 3 RO 3 RO 3 2 2 It is nearly two hundred years since the initial report the ubiquitous standard opium poppy or with cultivars that (1806) of the isolation of morphine (1, R = H) from the might be subsequently generated through genetic unripe seed pods of the opium poppy, Papever somniferum, manipulations so as to maximize production of these or by Friedrich Wihelm Adam Setürner [1], seventy five years related bases. -

DIENES Dienes Are Alkenes with 2 Double Bonds. IUPAC: Same As
DIENES Dienes are alkenes with 2 double bonds. IUPAC: Same as alkene, but change -ene to -adiene and use two numbers to locate the two double bonds (number from the end of the chain which makes the smaller of these numbers smaller). Double bonds separated by more than one single bond are isolated. Compounds with isolated double bonds have the same chemical properties as alkenes. Double bonds that alternate with single bonds, eg C=C-C=C, are conjugated. Double bond arrangements like C=C=C are cumulated. Compounds containing two carbon-carbon cumulated double bonds are called allenes. Conjugated dienes differ from simple alkenes in that they are more stable, undergo 1,4-addition, and are more reactive. Stability Alkene Type ∆ Hhydrogenation RCH=CH2 30 kcal/mole,exotherm. R2C=CH2, RCH=CHR 28 R2C=CHR 27 ∆ For dienes with isolated double bonds, the Hhydrogenation calculated by adding the appropriate values from the table above together is very close to the experimental value. ∆ For conjugated dienes, the experimental Hhydrogenation is smaller than the calculated one, which is based on isolated double bonds. Conjugated Diene Calculated Experimental ∆ ∆ Hh Hh CH2=CH-CH=CH2 60 kcal/mole 57 CH2=C(CH3)-C(CH3)=CH2 56 54 Therefore, conjugated dienes are more stable than isolated dienes. This stability is reflected in the fact that, where possible, conjugated dienes are the preferred diene products of elimination reactions --- H H C C C C H H H C C H C C KOH, alcohol major product - conjugated C C heat Cl H H H C + C C + H O + KCl Formation of the conjugated C C 2 diene takes precedence over H the Saytzeff rule.