Chem 51B Chapter 16 Notes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -

Brittain-DR-1965-Phd-Thesis.Pdf
POLYMETHYLENE PYRIDINES , A thesis submitted by David Robert Brittain in partial fulfilment of the requirements for the degree of DOCTOR OP PHILOSOPHY in the University of London Organic Chemistry Department, dune, 1965. Imperial College, LONDON, S.W.7. ABSTRACT This thesis describes a series of attempts to syn- thesise 2,5- and 1,4-polymethylene bridged pyridines. Nuclear magnetic resonance theory predicts that protons, which are held directly over an aromatic ring, will be abnormally shielded compared with protons in aliphatic straight-chain hydrocarbons. This prediction has been verified for the central methylene protons of paracyclo- phanes. The degree of shielding, expressed in terms of the distance from the aromatic ring, is a measure of the induced ring current and hence the aromaticity of the benzene ring. Similar measurements upon 2,5- or 1,4— polymethylene bridged pyridines would make it possible to determine the degree of aromaticity of the pyridine ring relative to benzene. A review of the subject of aromaticity is presented in which special reference has been made to its inter- pretation by nuclear magnetic resonance. The synthetic work has not been brougL.t to a truly satisfactory conclusion. However, the synthetic routes to 2,5-dialkylpyridines have been thoroughly investigated and a wide variety of such compounds prepared. The functional groups at the ends of the alkyl chains have been varied in an effort to produce a derivative which would cyclise to give a 2,5-bridged pyridine. The attempted intramolecular oxidative coupling of 2,5-dihex- 51 -ynylpyridine received much attention. In the attempts to obtain a 1,4-bridged pyridine, two tricyclic compounds, each containing two quaternised pyridine rings linked by polymethylene chains, were obtained. -

Chemical Kinetics HW1 (Kahn, 2010)
Chemical Kinetics HW1 (Kahn, 2010) Question 1. (6 pts) A reaction with stoichiometry A = P + 2Q was studied by monitoring the concentration of the reactant A as a function of time for eighteen minutes. The concentration determination method had a maximum error of 6 M. The following concentration profile was observed: Time (min) Conc (mM) 1 0.9850 2 0.8571 3 0.7482 4 0.6549 5 0.5885 6 0.5183 7 0.4667 8 0.4281 9 0.3864 10 0.3557 11 0.3259 12 0.3037 13 0.2706 14 0.2486 15 0.2355 16 0.2188 17 0.2111 18 0.1930 Determine the reaction order and calculate the rate constant for decomposition of A. What can be said about the mechanism or molecularity of this reaction? Question 2. (4 pts) Solve problem 2 on pg 31 in your textbook (House) using both linear and non-linear regression. Provide standard errors for the rate constant and half-life based on linear and non-linear fits. Below is the data set for your convenience: dataA = {{0, 0.5}, {10, 0.443}, {20,0.395}, {30,0.348}, {40,0.310}, {50,0.274}, {60,0.24}, {70,0.212}, {80,0.190}, {90,0.171}, {100,0.164}} Question 3. (3 pts) Solve problem 3 on pg 32 in your textbook (House). Question 4. (7 pts) The authors of the paper “Microsecond Folding of the Cold Shock Protein Measured by a Pressure-Jump Technique” suggest that the activated state of folding of CspB follows Hammond- type behavior. -

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

1St Year Students Lecture No. 5 Ahmed Mohamad Jawad
1st year students Lecture No. 5 Ahmed Mohamad Jawad Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Out line 1- Dienes: Introduction of diene Nomenclature of diene Preparation of diene Reaction of diene 2- Alkyne Introduction of Alkyne Nomenclature of Alkyne Physical properties of alkyne Preparation of Alkyne Reaction of Alkyne Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene Dienes are simply alkenes that contain two carbon-carbon double bonds. Dienes are divided into two major important classes according to the arrangement of the double bonds 1-Conjugated : Double bonds that alternate with single bonds are said to be conjugated. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene 2-Isolated double bonds that are separated by more than one single bond are said to be isolated Less stable than conjugated 3-Cumulated : contains cumulated double bonds Least one stable . Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Nomenclature of diene Dienes are named by the IUPAC system in the same way as alkenes , except that the ending diene is used, with two numbers to indicate the position of the two double bonds. H2C C CH2 1,2-propadiene 1,3-butadiene 1,4-pentadiene This system is easily extended to compounds containing any number of double bonds. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Preparation of diene 1- by acid catalyzed double dehydration 2- By dehydrogenation of dihalides: Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Reactions of Dienes In general terms, dienes undergo electrophilic addition reactions in a similar approach of alkenes Conjugated dienes undergo addition but the proximity of the conjugated C=C influences the reactions. -

Photoremovable Protecting Groups Used for the Caging of Biomolecules
1 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1 2-Nitrobenzyl and 7-Nitroindoline Derivatives John E.T. Corrie 1.1.1 Introduction 1.1.1.1 Preamble and Scope of the Review This chapter covers developments with 2-nitrobenzyl (and substituted variants) and 7-nitroindoline caging groups over the decade from 1993, when the author last reviewed the topic [1]. Other reviews covered parts of the field at a similar date [2, 3], and more recent coverage is also available [4–6]. This chapter is not an exhaustive review of every instance of the subject cages, and its principal focus is on the chemistry of synthesis and photocleavage. Applications of indi- vidual compounds are only briefly discussed, usually when needed to put the work into context. The balance between the two cage types is heavily slanted to- ward the 2-nitrobenzyls, since work with 7-nitroindoline cages dates essentially from 1999 (see Section 1.1.3.2), while the 2-nitrobenzyl type has been in use for 25 years, from the introduction of caged ATP 1 (Scheme 1.1.1) by Kaplan and co-workers [7] in 1978. Scheme 1.1.1 Overall photolysis reaction of NPE-caged ATP 1. Dynamic Studies in Biology. Edited by M. Goeldner, R. Givens Copyright © 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30783-4 2 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1.1.2 Historical Perspective The pioneering work of Kaplan et al. [7], although preceded by other examples of 2-nitrobenzyl photolysis in synthetic organic chemistry, was the first to apply this to a biological problem, the erythrocytic Na:K ion pump. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Diels-Alder Reaction Between Naphthalene and N-Phenylmaleimide Under Ambient and High Pressure Conditions
Issue in Honor of Prof. Alexander I. Konovalov ARKIVOC 2004 (xii) 70-79 Diels-Alder reaction between naphthalene and N-phenylmaleimide under ambient and high pressure conditions Gulnara G. Iskhakova*, Vladimir D. Kiselev, Elena A. Kashaeva, Lubov’ N. Potapova, Evgeny A. Berdnikov, Dmitry B. Krivolapov, and Igor A. Litvinov A. M. Butlerov Chemical Institute, Kazan State University, Kremlevskaya str.18, 420008 Kazan, Russian Federation E-mail: [email protected] Dedicated to the 70th anniversary of Professor Alexander I. Konovalov (received 02 Nov 04; accepted 08 Jan 05; published on the web 05 Feb 05) Abstract The rate and equilibrium constants for the Diels-Alder reactions between benzene and naphthalene as dienes and tetracyanoethylene, maleic anhydride and N-phenylmaleimide as dienophiles at 25 °С were estimated from empirical rule. The highest yield of the adduct was predicted for the reaction of naphthalene with N-phenylmaleimide. The time of adduct formation in 50% yield exceeds 30 years. The use of gallium chloride as a catalyst affords the exo-adduct for seven days at room temperature. The rate ((2±0.5)•10-6 L mol-1 s-1) and equilibrium constants (5±2 L mol-1) of this reaction were determined. Under high pressure conditions (8 kbar) reaction occurs with formation of both stereo isomers at 100 °С during 80 hours. Keywords: Naphthalene, Diels-Alder reaction, catalysis, high pressure Introduction 1 Many dienes, including substituted benzenes and naphthalenes, are known to form molecular complexes of the π,π-type due to the interaction of the highest occupied π-orbital of a diene- donor with the lowest unoccupied π-orbital of a dienophile-acceptor. -

Reaction Mechanism Lecture 3 : Nucleophilic And
Module 10 : Reaction mechanism Lecture 3 : Nucleophilic and Electrophilic Addition and Abstraction Objectives In this lecture you will learn the following Ligand activation by metal that leads to a direct external attack at the ligand. Nucleophilic addition and nucleophilic abstraction reactions. Electrophilic addition and electrophilic abstraction reactions. The nucleophilic and electrophilic substitution and abstraction reactions can be viewed as ways of activation of substrates to allow an external reagent to directly attack the metal activated ligand without requiring prior binding of the external reagent to the metal. The attacking reagent may be a nucleophile or an electrophile. The nucleophilic attack of the external reagent is favored if the LnM fragment is a poor π−base and a good σ−acid i.e., when the complex is cationic and/or when the other metal bound ligands are electron withdrawing such that the ligand getting activated gets depleted of electron density and can undergo an external attack by a nucleophile Nu−, like LiMe or OH−. The attack of the nucleophiles may result in the formation of a bond between the nucleophiles and the activated unsaturated substrate, in which case it is called nucleophilic addition, or may result in an abstraction of a part or the whole of the activated ligand, in which case it is called the nucleophilic abstraction. The nucleophilic addition and the abstraction reactions are discussed below. Nucleophilic addition An example of a nucleophilic addition reaction is shown below. Carbon monoxide (CO) as a ligand can undergo nucleophilic attack when bound to a metal center of poor π−basicity, as the carbon center of the CO ligand is electron deficient owing to the ligand to metal σ−donation not being fully compensated by the metal to ligand π−back donation. -
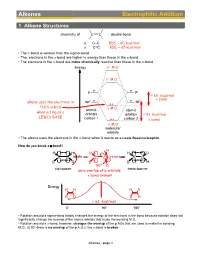
Alkenes Electrophilic Addition
Alkenes Electrophilic Addition 1 Alkene Structures chemistry of C C double bond σ C–C BDE ~ 80 kcal/mol π C=C BDE ~ 65 kcal/mol • The p-bond is weaker than the sigma-bond • The, electrons in the p-bond are higher in energy than those in the s-bond • The electrons in the p-bond are more chemically reactive than those in the s-bond Energy σ∗ M.O. π∗ M.O. p p ~ 65 kcal/mol 2 π bond alkene uses the electrons in sp2 sp THIS orbital π M.O. atomic atomic when acting as a orbitals orbitals ~ 81 kcal/mol LEWIS BASE carbon 1 carbon 2 σ bond σ M.O. molecular orbitals • The alkene uses the electrons in the p-bond when it reacts as a Lewis Base/nucleophile How do you break a p-bond? H H H H H D C C rotate C C rotate C C D D D D H 90° D cis-isomer trans-isomer zero overlap of p orbitals: π bond broken! Energy H H H D D D D H ~ 63 kcal/mol 0° 90° 180° • Rotation around a sigma-bond hardly changes the energy of the electrons in the bond because rotation does not significantly change the overlap of the atomic orbitals that make the bonding M.O. • Rotation around a p-bond, however, changes the overlap of the p AOs that are used to make the bonding M.O., at 90° there is no overlap of the p A.O.s, the p-bond is broken Alkenes : page 1 Distinguishing isomers trans- cis- • By now we are very familiar with cis- and trans-stereoisomers (diastereomers) • But what about, the following two structures, they can NOT be assigned as cis- or trans-, yet they are definitely stereoisomers (diastereomers), the directions in which their atoms point in space are different cis- / trans- Br Br We Need a different system to distinguish stereoisomers for C=C double bonds: Use Z/E notation. -

Subject Index
455 Subject Index Aminohydroxylation, 364 a-Aminoketone, 281 4-Aminophenol, 18 A Aminothiophene, 158 Abnormal Claisen rearrangement, 1 a-Aminothiophenols, 184 Acrolein, 378 Ammonium ylide, 383 2-(Acylamino)-toluenes, 245 Angeli-Rimini hydroxamic acid Acylation, 100, 145, 200 synthesis, 9 Acyl azides, 98 ~-Anomer, 225 Acylium ion, 145, 149, 175, 177 Anomeric center, 211 a-Acyloxycarboxamides, 298 Anomeric effect, 135 a-Acyloxyketones, 17 ANRORC mechanism, 10 a-Acyloxythioethers, 327 Anthracenes, 51 Acyl transfer, 17, 42, 228, 298, 305, Anti-Markovnikov addition, 219 327,345 Amdt-Eistert homologation, 11 AIBN, 22, 23, 415 Aryl-acetylene, 66 Alder ene reaction, 2 Arylation, 253 Alder's endo rule, Ill 0-Aryliminoethers, 67 Aldol condensation, 3, 14, 26, 34, 2-Arylindoles, 38 69, 130, 147, 172, 305, Aryl migration, 31 340,396,412 Autoxidation, 69, 115, 118 Aldosylamine, 8 Auwers reaction, 13 Alkyl migration, 16, 132, 315, 443 Axial, 347 Alkylation, 144, 145 Azalactone, I 00 N-Aikylation, 162 Azides, 125, 330 Alkylidene carbene, 151 Azirine, 6, 7, 281 Allan-Robinson reaction, 4, 228 Azulene, 310 Allene, 119 1t-Allyl complex, 414 B Allylation, 213, 414 Baeyer-Drewson Allylstannane, 213 indigo synthesis, 14 Allylsilanes, 349 Baeyer-Villiger oxidation, 16, 53 Alper carbonylation, 6 Baker-Venkataraman Alpine-borane®, 262 rearrangement, 17 Aluminum phenolate, 149 Balz-Schiemann reaction, 354 Amadori rearrangement, 8 Bamberger rearrangement, 18 Amide acetal, 74 Bamford-Stevens reaction, 19 Amides, 28, 67,276,339, 356 Bargellini reaction, 20 Amidine, -
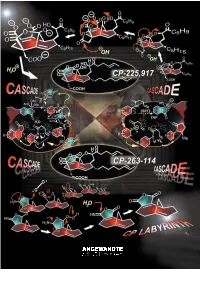
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis.