Protecting Groups Are Attacked by the by Attacked Are Groups Protecting -Troc -Phthalimido), Benzylidene and Isopropyl- O N CO 2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brittain-DR-1965-Phd-Thesis.Pdf
POLYMETHYLENE PYRIDINES , A thesis submitted by David Robert Brittain in partial fulfilment of the requirements for the degree of DOCTOR OP PHILOSOPHY in the University of London Organic Chemistry Department, dune, 1965. Imperial College, LONDON, S.W.7. ABSTRACT This thesis describes a series of attempts to syn- thesise 2,5- and 1,4-polymethylene bridged pyridines. Nuclear magnetic resonance theory predicts that protons, which are held directly over an aromatic ring, will be abnormally shielded compared with protons in aliphatic straight-chain hydrocarbons. This prediction has been verified for the central methylene protons of paracyclo- phanes. The degree of shielding, expressed in terms of the distance from the aromatic ring, is a measure of the induced ring current and hence the aromaticity of the benzene ring. Similar measurements upon 2,5- or 1,4— polymethylene bridged pyridines would make it possible to determine the degree of aromaticity of the pyridine ring relative to benzene. A review of the subject of aromaticity is presented in which special reference has been made to its inter- pretation by nuclear magnetic resonance. The synthetic work has not been brougL.t to a truly satisfactory conclusion. However, the synthetic routes to 2,5-dialkylpyridines have been thoroughly investigated and a wide variety of such compounds prepared. The functional groups at the ends of the alkyl chains have been varied in an effort to produce a derivative which would cyclise to give a 2,5-bridged pyridine. The attempted intramolecular oxidative coupling of 2,5-dihex- 51 -ynylpyridine received much attention. In the attempts to obtain a 1,4-bridged pyridine, two tricyclic compounds, each containing two quaternised pyridine rings linked by polymethylene chains, were obtained. -

IV (Advance Organic Synthesis and Supramolecular Chemistry and Carbocyclic Rings) Module No and 9: Protection and Deprotection Title Module Tag CHE P14 M9
Subject Chemistry Paper No and Title 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No and 9: Protection and deprotection Title Module Tag CHE_P14_M9 CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 9: Protection and deprotection Table of Content 1. Learning outcomes 2. Introduction 3. Protecting groups for carbonyl compounds 4. Protecting groups for alcohols 5. Protecting groups for amines 6. Summary CHEMISTRY Paper No. 14: Organic Chemistry –IV (Advance Organic Synthesis and Supramolecular Chemistry and carbocyclic rings) Module No. 9: Protection and deprotection 1. Learning Outcomes After studying this module, you shall be able to Know about protecting groups. Study the characteristics of protecting groups. Understand the functional group protection. Know the protection of important functional groups. 2. Introduction Protection and deprotection is an important part of organic synthesis. During the course of synthesis, we many times desire to perform reaction at only one of the two functional groups in any single organic molecules. For example, in an organic compound possessing two functional groups like ester and ketone, we have to perform reaction at only ester group, them the keto group needs to be protected. If we want to reduce the ester group, then keto group will also get reduced. To avoid this type of complications, protection and deprotection of functional groups are necessary. 3. Protecting groups for carbonyl compounds The protecting groups allow the masking of a particular functional group where a specified reaction is not to be performed. The protection is required as it interferes with another reaction. -

Qualitative Microanalysis of Non-Ferrous Metals
Proceedings of the Iowa Academy of Science Volume 49 Annual Issue Article 53 1942 Qualitative Microanalysis of Non-Ferrous Metals George W. Brown University of Iowa Lothrop Smith University of Iowa Let us know how access to this document benefits ouy Copyright ©1942 Iowa Academy of Science, Inc. Follow this and additional works at: https://scholarworks.uni.edu/pias Recommended Citation Brown, George W. and Smith, Lothrop (1942) "Qualitative Microanalysis of Non-Ferrous Metals," Proceedings of the Iowa Academy of Science, 49(1), 323-331. Available at: https://scholarworks.uni.edu/pias/vol49/iss1/53 This Research is brought to you for free and open access by the Iowa Academy of Science at UNI ScholarWorks. It has been accepted for inclusion in Proceedings of the Iowa Academy of Science by an authorized editor of UNI ScholarWorks. For more information, please contact [email protected]. Brown and Smith: Qualitative Microanalysis of Non-Ferrous Metals QUALITATIVE MICROANALYSIS OF NON-FERROUS METALS GEOIWE w. BROWN AND LOTHROP SMITH During summer vacations, part of my school expense was earn ed as a purchaser of non-ferrous scrap metal for a small Chicago smelter. In such purchasing, it is very necessary to know some thing of the general composition of the metal. For example, in buying soldered articles, it is of vital importance to know whether the solder is high in tin or lead, and whether it contains antimony or bismuth. Other purchasers to whom I have talked have report~ ed the same difficulty. It is common practice in the purchase of metal from large smelt ers, to take the vendor's word for the composition, as the only al ternatives are either an expensive analysis by a commercial labor atory, or the establishment of a laboratory by the small foundry. -

Syntheses and Eliminations of Cyclopentyl Derivatives David John Rausch Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Syntheses and eliminations of cyclopentyl derivatives David John Rausch Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Rausch, David John, "Syntheses and eliminations of cyclopentyl derivatives " (1966). Retrospective Theses and Dissertations. 2875. https://lib.dr.iastate.edu/rtd/2875 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 66—6996 RAUSCH, David John, 1940- SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES. Iowa State University of Science and Technology Ph.D., 1966 Chemistry, organic University Microfilms, Inc., Ann Arbor, Michigan SYNTHESES AND ELIMINATIONS OF CYCLOPENTYL DERIVATIVES by David John Rausch A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Organic Chemistry Approved : Signature was redacted for privacy. Signature was redacted for privacy. Head of Major Department Signature was redacted for privacy. Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OF CONTENTS VITA INTRODUCTION HISTORICAL Conformation of Cyclopentanes Elimination Reactions RESULTS AND DISCUSSION Synthetic Elimination Reactions EXPERIMENTAL Preparation and Purification of Materials Procedures and Data for Beta Elimination Reactions SUMMARY LITERATURE CITED ACKNOWLEDGEMENTS iii VITA The author was born in Aurora, Illinois, on October 24, 1940, to Mr. -

Synthesis of New Peptide Mimetics
University of Bath PHD Synthesis of new peptide mimetics Hackett, Anne Award date: 1996 Awarding institution: University of Bath Link to publication Alternative formats If you require this document in an alternative format, please contact: [email protected] General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Download date: 07. Oct. 2021 Synthesis of New Peptide Mimetics Submitted by Anne Hackett for the degree of PhD of the University of Bath 1996 COPYRIGHT Attention is drawn to the fact that copyright of this thesis rests with its author. This copy of the thesis has been supplied on condition that anyone who consults it is understood to recognise that its copyright rests with its author and that no quotation from the thesis and no information derived from it may be published without the prior written consent of the author. This thesis may not be consulted, photocopied or lent to other libraries without the permission of the author from three years from the date of acceptance of the thesis. -

1St Year Students Lecture No. 5 Ahmed Mohamad Jawad
1st year students Lecture No. 5 Ahmed Mohamad Jawad Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Out line 1- Dienes: Introduction of diene Nomenclature of diene Preparation of diene Reaction of diene 2- Alkyne Introduction of Alkyne Nomenclature of Alkyne Physical properties of alkyne Preparation of Alkyne Reaction of Alkyne Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene Dienes are simply alkenes that contain two carbon-carbon double bonds. Dienes are divided into two major important classes according to the arrangement of the double bonds 1-Conjugated : Double bonds that alternate with single bonds are said to be conjugated. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Introduction of diene 2-Isolated double bonds that are separated by more than one single bond are said to be isolated Less stable than conjugated 3-Cumulated : contains cumulated double bonds Least one stable . Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Nomenclature of diene Dienes are named by the IUPAC system in the same way as alkenes , except that the ending diene is used, with two numbers to indicate the position of the two double bonds. H2C C CH2 1,2-propadiene 1,3-butadiene 1,4-pentadiene This system is easily extended to compounds containing any number of double bonds. Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Preparation of diene 1- by acid catalyzed double dehydration 2- By dehydrogenation of dihalides: Create PDF files without this message by purchasing novaPDF printer (http://www.novapdf.com) Reactions of Dienes In general terms, dienes undergo electrophilic addition reactions in a similar approach of alkenes Conjugated dienes undergo addition but the proximity of the conjugated C=C influences the reactions. -

Photoremovable Protecting Groups Used for the Caging of Biomolecules
1 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1 2-Nitrobenzyl and 7-Nitroindoline Derivatives John E.T. Corrie 1.1.1 Introduction 1.1.1.1 Preamble and Scope of the Review This chapter covers developments with 2-nitrobenzyl (and substituted variants) and 7-nitroindoline caging groups over the decade from 1993, when the author last reviewed the topic [1]. Other reviews covered parts of the field at a similar date [2, 3], and more recent coverage is also available [4–6]. This chapter is not an exhaustive review of every instance of the subject cages, and its principal focus is on the chemistry of synthesis and photocleavage. Applications of indi- vidual compounds are only briefly discussed, usually when needed to put the work into context. The balance between the two cage types is heavily slanted to- ward the 2-nitrobenzyls, since work with 7-nitroindoline cages dates essentially from 1999 (see Section 1.1.3.2), while the 2-nitrobenzyl type has been in use for 25 years, from the introduction of caged ATP 1 (Scheme 1.1.1) by Kaplan and co-workers [7] in 1978. Scheme 1.1.1 Overall photolysis reaction of NPE-caged ATP 1. Dynamic Studies in Biology. Edited by M. Goeldner, R. Givens Copyright © 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30783-4 2 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1.1.2 Historical Perspective The pioneering work of Kaplan et al. [7], although preceded by other examples of 2-nitrobenzyl photolysis in synthetic organic chemistry, was the first to apply this to a biological problem, the erythrocytic Na:K ion pump. -

Diels-Alder Reaction Between Naphthalene and N-Phenylmaleimide Under Ambient and High Pressure Conditions
Issue in Honor of Prof. Alexander I. Konovalov ARKIVOC 2004 (xii) 70-79 Diels-Alder reaction between naphthalene and N-phenylmaleimide under ambient and high pressure conditions Gulnara G. Iskhakova*, Vladimir D. Kiselev, Elena A. Kashaeva, Lubov’ N. Potapova, Evgeny A. Berdnikov, Dmitry B. Krivolapov, and Igor A. Litvinov A. M. Butlerov Chemical Institute, Kazan State University, Kremlevskaya str.18, 420008 Kazan, Russian Federation E-mail: [email protected] Dedicated to the 70th anniversary of Professor Alexander I. Konovalov (received 02 Nov 04; accepted 08 Jan 05; published on the web 05 Feb 05) Abstract The rate and equilibrium constants for the Diels-Alder reactions between benzene and naphthalene as dienes and tetracyanoethylene, maleic anhydride and N-phenylmaleimide as dienophiles at 25 °С were estimated from empirical rule. The highest yield of the adduct was predicted for the reaction of naphthalene with N-phenylmaleimide. The time of adduct formation in 50% yield exceeds 30 years. The use of gallium chloride as a catalyst affords the exo-adduct for seven days at room temperature. The rate ((2±0.5)•10-6 L mol-1 s-1) and equilibrium constants (5±2 L mol-1) of this reaction were determined. Under high pressure conditions (8 kbar) reaction occurs with formation of both stereo isomers at 100 °С during 80 hours. Keywords: Naphthalene, Diels-Alder reaction, catalysis, high pressure Introduction 1 Many dienes, including substituted benzenes and naphthalenes, are known to form molecular complexes of the π,π-type due to the interaction of the highest occupied π-orbital of a diene- donor with the lowest unoccupied π-orbital of a dienophile-acceptor. -
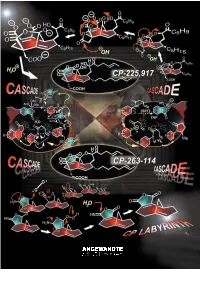
A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis
REVIEWS The CP Molecule Labyrinth: A Paradigm of How Endeavors in Total Synthesis Lead to Discoveries and Inventions in Organic Synthesis K. C. Nicolaou* and Phil S. Baran Dedicated to Mrs. Niki Goulandris for her outstanding contributions to humanity and Planet Earth on the occasion of the opening of the GAIA Center for Environmental Research and Education at the Goulandris Natural History Museum in Athens, Greece. Imagine an artist carving a sculpture Herculean nature of the task and the ed Minotaur, which he accomplished from a marble slab and finding gold rewards that accompany it, one must through brilliance, skill, and bravery nuggets in the process. This thought is sense the details of the enterprise having traversed the famous labyrinth not a far-fetched description of the behind the scenes. A more vivid de- with the help of Ariadne. This story work of a synthetic chemist pursuing scription of total synthesis as a struggle from Greek mythology comes alive in the total synthesis of a natural product. against a tough opponent is perhaps modern synthetic expeditions toward At the end of the day, he or she will be appropriate to dramatize these ele- natural products as exemplified by the judged by the artistry of the final work ments of the experience. In this article total synthesis of the CP molecules and the weight of the gold discovered we describe one such endeavor of total which serve as a paradigm for modern in the process. However, as colorful as synthesis which, in addition to reaching total synthesis endeavors, where the this description of total synthesis may the target molecule, resulted in a objectives are discovery and invention be, it does not entirely capture the wealth of new synthetic strategies and in the broader sense of organic syn- essence of the endeavor, for there is technologies for chemical synthesis. -

Chapter 3. the Concept of Protecting Functional Groups
Chapter 3. The Concept of Protecting Functional Groups When a chemical reaction is to be carried out selectively at one reactive site in a multifunctional compound, other reactive sites must be temporarily blocked. A protecting group must fulfill a number of requirements: • The protecting group reagent must react selectively (kinetic chemoselectivity) in good yield to give a protected substrate that is stable to the projected reactions. • The protecting group must be selectively removed in good yield by readily available reagents. • The protecting group should not have additional functionality that might provide additional sites of reaction. 3.1 Protecting of NH groups Primary and secondary amines are prone to oxidation, and N-H bonds undergo metallation on exposure to organolithium and Grignard reagents. Moreover, the amino group possesses a lone pair electrons, which can be protonated or reacted with electrophiles. To render the lone pair electrons less reactive, the amine can be converted into an amide via acylation. N-Benzylamine Useful for exposure to organometallic reagents or metal hydrides Hydrogenolysis Benzylamines are not cleaved by Lewis acid Pearlman’s catalyst Amides Basicity of nitrogen is reduced, making them less susceptible to attack by electrophilic reagent The group is stable to pH 1-14, nucleophiles, organometallics (except organolithium reagents), catalytic hydrogenation, and oxidation. Cleaved by strong acid (6N HCl, HBr) or diisobutylaluminum hydride Carbamates Behave like a amides, hence no longer act as nucleophile Stable to oxidizing agents and aqueous bases but may react with reducing agents. Iodotrimethylsilane is often the reagent for removal of this protecting group Stable to both aqueous acid and base Benzoyloxycarbonyl group for peptide synthesis t-butoxycarbonyl group(Boc) is inert to hydrogenolysis and resistant to bases and nucleophilic reagent. -

Disconnect by the Numbers: a Beginner's Guide to Synthesis
Disconnect by the Numbers A Beginner’s Guide to Synthesis Michael B. Smith University of Connecticut, Storrs, CT 06269 A perennial problem in teaching organic chemistry is in- countered in undergraduate courses, however, quickly re- troduction of synthesis and the disconnection method. veals that such complexity is lacking in most cases and a Sophomores in the midst of the first organic course and first- simple bond-forming strategy is both reasonable and useful. year graduate students with a single undergraduate organic The protocol described in this work will begin by assigning course have only a limited knowledge of carbon bond form- numbers to each bond, based entirely on the small set of ing reactions and functional group interchange reactions. carbon bond forming reactions introduced in a typical two- The student often worries more about making the “correct semester organic chemistry course (7). These reactions are disconnection” rather than focusing attention on the chemi- listed in Table 1. Tables are provided to assign the numbers, cal reactions and concepts required to form that bond. When based on the importance of forming carbon bonds in mole- the student makes a reasonable disconnection, the choices to cules containing a polarized functional group. The proce- reform that bond are often limited and confusing. dure assigns priorities based on key regions of a molecule: (1) The synthesis of organic molecules dates to the 19th cen- the bond connected to a functional group X (C—/ /—X), (2) tury, but the work of Perkin, Robinson, and others in the to the second bond from X (C—//—C—X) and (3) to the early 20th century demonstrated the ability to plan a syn- third bond from X (C—/ /—C—C—X). -

Industrial Chemistry Process Scheme for Sections 3 - 3.3.2 Butenes Natural Gas Lwet I Oil
Olefins - Part 1 Industrial Chemistry Process Scheme for Sections 3 - 3.3.2 Butenes Natural Gas lwet I Oil Gas Gas Naphtha Oil Vacuum Distillation iiHydrotreating Naphtha Catalytic and Steam Thermal Crack- Process &--Crac kin ------l Where they come from Ethylenenm Propene Acetylene Ethylene Fuel Gas Ethylene Propene CL- C5- Pyrolysis Fraction Fraction Gasoline , + I 112.2.1 CL-Riffinate C5- Raffinate Ethylene 2-Butene manufacture Ratfinate I1 isobutene or n- Butenes n- and lsopentenes n-Pentenes lsobutene Oligomers n- and lsobutane n- and lsopentanes n- and lsopentanes 68 3. Olejins most valuable C4 olefin source: As shown in Table 3-3, a significantly higher fraction of steam cracking of naphtha butenes is obtained from steam cracking of naphtha than from Which C4 are obtainedcatalytic by cracking steam of gas oil. Therefore cracking? naphtha steam cracking is the more interesting technology for production of unsatu- rated C4 compounds. decrease of total C4 and C4 olefins, but As the cracking- severity increases, both the total yield of the increase Of butadiene under high severity C4 fraction and the proportion of butenes decrease, while the cracking conditions (consequence of butanedesired higher C2H4isobutane yield) proportion of butadiene increases due to its higher stability: Table 3-3. Composition of Cq fractions from steam cracking of naphtha and catalytic cracking of gas oil (in wt%). but-1-ene (E)-but-2-ene Steam cracking Catalytic cracking 1-butene Cracked products Low High (FCC) zeolite trans-2-butene severity seventy catalyst