Mutation and LOH Analysis of ACO2 in Colorectal Cancer: No Evidence Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -
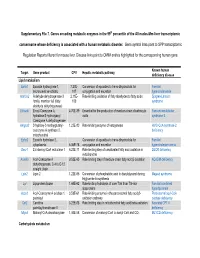
Supplementary File 7. Genes Encoding Metabolic Enzymes in The
Supplementary File 7. Genes encoding metabolic enzymes in the 99th percentile of the All nodes-Mm-liver transcriptomic consensome whose deficiency is associated with a human metabolic disorder. Gene symbol links point to SPP transcriptomic Regulation Reports filtered for mouse liver. Disease links point to OMIM entries highlighted for the corresponding human gene. Known human Target Gene product CPV Hepatic metabolic pathway deficiency disease Lipid metabolism Ephx1 Epoxide hydroxylase 1, 7.27E- Conversion of epoxides to trans-dihydrodiols for Familial microsomal xenobiotic 107 conjugation and excretion hypercholanemia Aldh3a2 Aldehyde dehydrogenase 3 2.11E- Rate-limiting oxidation of fatty aldehydes to fatty acids Sjogren-Larsson family, member A2 (fatty 103 syndrome aldehyde dehydrogenase) Ehhadh Enoyl-Coenzyme A, 4.70E-89 Essential for the production of medium-chain dicarboxylic Fanconi renotubular hydratase/3-hydroxyacyl acids syndrome 3 Coenzyme A dehydrogenase Hmgcs2 3-hydroxy-3-methylglutaryl- 1.23E-80 Rate-limiting enzyme of ketogenesis HMG-CoA synthase-2 coenzyme A synthase 2, deficiency mitochondrial Ephx2 Epoxide hydrolase 2, Conversion of epoxides to trans-dihydrodiols for Familial cytoplasmic 6.06E-78 conjugation and excretion hypercholesterolemia Decr1 2,4-dienoyl-CoA reductase 1 6.23E-71 Rate-limiting step of unsaturated fatty acid oxidation in DECR deficiency mitochondria Acadm Acyl-Coenzyme A 9.52E-65 Rate limiting step of medium-chain fatty acid β-oxidation ACADM deficiency dehydrogenase, C-4 to C-12 straight chain Lpin2 -

Complex Hereditary Spastic Paraplegia Associated with Episodic
Tozawa et al. Human Genome Variation (2021) 8:4 https://doi.org/10.1038/s41439-021-00136-y Human Genome Variation DATA REPORT Open Access Complex hereditary spastic paraplegia associated with episodic visual loss caused by ACO2 variants Takenori Tozawa1,2,AkiraNishimura3, Tamaki Ueno2,4, Akane Shikata5, Yoshihiro Taura1,TakeshiYoshida 6, Naoko Nakagawa7, Takahito Wada 7, Shinji Kosugi7, Tomoko Uehara8, Toshiki Takenouchi 9, Kenjiro Kosaki8 and Tomohiro Chiyonobu1 Abstract Most patients with homozygous or compound heterozygous pathogenic ACO2 variants present with muscular hypotonia features, namely, infantile cerebellar-retinal degeneration. Recently, two studies reported rare familial cases of ACO2 variants presenting as complex hereditary spastic paraplegia (HSP) with broad clinical spectra. Here, we report the case of a 20-year-old Japanese woman with complex HSP caused by compound heterozygous ACO2 variants, revealing a new phenotype of episodic visual loss during febrile illness. The ACO2 gene on chromosome 22 encodes the aco- variants in the ACO2 gene presenting as complex her- nitase 2 (ACO2) protein in the mitochondrial matrix; editary spastic paraplegia (HSP) with a new phenotype of ACO2 catalyzes the stereospecific isomerization of citrate episodic visual loss after every febrile infection and pro- to isocitrate in the tricarboxylic acid (TCA) cycle1. gressive optic atrophy. This is the third familial report and ACO2 fi fi 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Pathogenic variants were rst reported in eight the rst Asian patient with complex HSP caused by individuals from two Arab families, and they had infantile pathogenic ACO2 variants. cerebellar-retinal degeneration (ICRD, OMIM#614559)2. The proband was born to nonconsanguineous healthy Subsequently, ~20 cases of pathogenic homozygous or parents at 38 weeks gestational age after unremarkable compound heterozygous ACO2 variants have been delivery. -
![(ACO2) Mouse Monoclonal Antibody [Clone ID: OTI7G4] Product Data](https://docslib.b-cdn.net/cover/3055/aco2-mouse-monoclonal-antibody-clone-id-oti7g4-product-data-823055.webp)
(ACO2) Mouse Monoclonal Antibody [Clone ID: OTI7G4] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA500873 Aconitase 2 (ACO2) Mouse Monoclonal Antibody [Clone ID: OTI7G4] Product data: Product Type: Primary Antibodies Clone Name: OTI7G4 Applications: FC, IF, WB Recommended Dilution: WB 1:2000, IF 1:100, Flow 1:100 Reactivity: Human, Mouse, Rat Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human ACO2 (NP_001089) produced in HEK293T cell. Formulation: PBS (pH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 85.4 kDa Gene Name: aconitase 2 Database Link: NP_001089 Entrez Gene 11429 MouseEntrez Gene 79250 RatEntrez Gene 50 Human Q99798 Background: The protein encoded by this gene belongs to the aconitase/IPM isomerase family. It is an enzyme that catalyzes the interconversion of citrate to isocitrate via cis-aconitate in the second step of the TCA cycle. This protein is encoded in the nucleus and functions in the mitochondrion. It was found to be one of the mitochondrial matrix proteins that are preferentially degraded by the serine protease 15(PRSS15), also known as Lon protease, after oxidative modification. This product is to be used for laboratory only. Not for diagnostic or therapeutic use. -

Hepatic Cystathionine Β-Synthase Activity Is Increased by Greater Postruminal Supply of Met During the Periparturient Period in Dairy Cows
ORIGINAL RESEARCH CURRENT DEVELOPMENTS IN NUTRITION Nutritional Biochemistry and Physiologic Mechanisms Hepatic Cystathionine β-Synthase Activity Is Increased by Greater Postruminal Supply of Met during the Periparturient Period in Dairy Cows Mario Vailati-Riboni,1 Fernanda Batistel,1 Rainie RCS Yambao,1 Claudia Parys,2 Yuan-Xiang Pan,3 and Juan J Loor1 1Department of Animal Sciences and Division of Nutritional Sciences, University of Illinois, Urbana, IL, USA; 2Evonik Nutrition & Care GmbH, Hanau-Wolfgang, Germany; and 3Department of Food Science and Human Nutrition and Division of Nutritional Sciences, University of Illinois, Urbana, IL, USA ABSTRACT Background: Postruminal supply of Met during the periparturient period enhances production efficiency (feed conversion to milk) in dairy cows partly through alleviation of oxidant and inflammatory status. Whether alterations in hepatic 1-carbon metabolism (major contributor of antioxidants) and/or energy metabolism contribute to these beneficial effects is unknown. Objectives: To investigate alterations in hepatic 1-carbon and energy metabolism and associations with plasma amino acids (AAs) and production efficiency in response to enhanced postruminal supply of Met. Methods: Holstein cows (n = 30 per group) were fed during the last 28 d of pregnancy a control diet (CON) or the control plus ethylcellulose rumen-protected Met (MET; 0.9 g/kg of dry matter intake). Plasma (n = 15 per group) and liver tissue (n = 10 per group) were collected throughout the periparturient period to evaluate AA profiles, activity of the tricarboxylic acid cycle, and 1-carbon metabolism via mRNA abundance, enzyme activity, and targeted metabolomics. Results: Cows in the MET group had greater overall (27%, P = 0.027) plasma Met concentrations, but had similar total plasma AA concentrations. -

Whole Exome Sequencing Gene Package Intellectual Disability, Version 9.1, 31-1-2020
Whole Exome Sequencing Gene package Intellectual disability, version 9.1, 31-1-2020 Technical information DNA was enriched using Agilent SureSelect DNA + SureSelect OneSeq 300kb CNV Backbone + Human All Exon V7 capture and paired-end sequenced on the Illumina platform (outsourced). The aim is to obtain 10 Giga base pairs per exome with a mapped fraction of 0.99. The average coverage of the exome is ~50x. Duplicate and non-unique reads are excluded. Data are demultiplexed with bcl2fastq Conversion Software from Illumina. Reads are mapped to the genome using the BWA-MEM algorithm (reference: http://bio-bwa.sourceforge.net/). Variant detection is performed by the Genome Analysis Toolkit HaplotypeCaller (reference: http://www.broadinstitute.org/gatk/). The detected variants are filtered and annotated with Cartagenia software and classified with Alamut Visual. It is not excluded that pathogenic mutations are being missed using this technology. At this moment, there is not enough information about the sensitivity of this technique with respect to the detection of deletions and duplications of more than 5 nucleotides and of somatic mosaic mutations (all types of sequence changes). HGNC approved Phenotype description including OMIM phenotype ID(s) OMIM median depth % covered % covered % covered gene symbol gene ID >10x >20x >30x A2ML1 {Otitis media, susceptibility to}, 166760 610627 66 100 100 96 AARS1 Charcot-Marie-Tooth disease, axonal, type 2N, 613287 601065 63 100 97 90 Epileptic encephalopathy, early infantile, 29, 616339 AASS Hyperlysinemia, -

Psykisk Utviklingshemming Og Forsinket Utvikling
Psykisk utviklingshemming og forsinket utvikling Genpanel, versjon v03 Tabellen er sortert på gennavn (HGNC gensymbol) Navn på gen er iht. HGNC >x10 Andel av genet som har blitt lest med tilfredstillende kvalitet flere enn 10 ganger under sekvensering x10 er forventet dekning; faktisk dekning vil variere. Gen Gen (HGNC Transkript >10x Fenotype (symbol) ID) AAAS 13666 NM_015665.5 100% Achalasia-addisonianism-alacrimia syndrome OMIM AARS 20 NM_001605.2 100% Charcot-Marie-Tooth disease, axonal, type 2N OMIM Epileptic encephalopathy, early infantile, 29 OMIM AASS 17366 NM_005763.3 100% Hyperlysinemia OMIM Saccharopinuria OMIM ABCB11 42 NM_003742.2 100% Cholestasis, benign recurrent intrahepatic, 2 OMIM Cholestasis, progressive familial intrahepatic 2 OMIM ABCB7 48 NM_004299.5 100% Anemia, sideroblastic, with ataxia OMIM ABCC6 57 NM_001171.5 93% Arterial calcification, generalized, of infancy, 2 OMIM Pseudoxanthoma elasticum OMIM Pseudoxanthoma elasticum, forme fruste OMIM ABCC9 60 NM_005691.3 100% Hypertrichotic osteochondrodysplasia OMIM ABCD1 61 NM_000033.3 77% Adrenoleukodystrophy OMIM Adrenomyeloneuropathy, adult OMIM ABCD4 68 NM_005050.3 100% Methylmalonic aciduria and homocystinuria, cblJ type OMIM ABHD5 21396 NM_016006.4 100% Chanarin-Dorfman syndrome OMIM ACAD9 21497 NM_014049.4 99% Mitochondrial complex I deficiency due to ACAD9 deficiency OMIM ACADM 89 NM_000016.5 100% Acyl-CoA dehydrogenase, medium chain, deficiency of OMIM ACADS 90 NM_000017.3 100% Acyl-CoA dehydrogenase, short-chain, deficiency of OMIM ACADVL 92 NM_000018.3 100% VLCAD -

(ACO2) Deficiency
RESEARCH ARTICLE Plasma metabolomics reveals a diagnostic metabolic fingerprint for mitochondrial aconitase (ACO2) deficiency Lucia Abela1,2,3, Ronen Spiegel4, Lisa M. Crowther1,2,3, Andrea Klein1¤a, Katharina Steindl3,5, Sorina Mihaela Papuc3,5, Pascal Joset3,5, Yoav Zehavi4, Anita Rauch3,5, Barbara Plecko1,2,3, Thomas Luke Simmons1,2,3¤b* 1 Division of Child Neurology, University Children's Hospital Zurich, Zurich, Switzerland, 2 Children's Research Centre, University Children's Hospital Zurich, Zurich, Switzerland, 3 Radiz±Rare Disease Initiative a1111111111 Zurich, Clinical Research Priority Program for Rare Diseases, University of Zurich, Zurich, Switzerland, a1111111111 4 Department of Pediatrics B, Emek Medical Center, Afula, Rappaport Faculty of Medicine, Technion, Israel, a1111111111 5 Institute of Medical Genetics, University of Zurich, Schlieren, Switzerland a1111111111 a1111111111 ¤a Current address: Pediatric Neurology, University of Basel Children's Hospital, Basel, Switzerland ¤b Current address: Evolva AG, Reinach, Switzerland * [email protected] OPEN ACCESS Abstract Citation: Abela L, Spiegel R, Crowther LM, Klein A, Mitochondrial respiratory chain dysfunction has been identified in a number of neurodegen- Steindl K, Papuc SM, et al. (2017) Plasma metabolomics reveals a diagnostic metabolic erative disorders. Infantile cerebellar-retinal degeneration associated with mutations in the fingerprint for mitochondrial aconitase (ACO2) mitochondrial aconitase 2 gene (ACO2) has been recently described as a neurodegenera- deficiency. PLoS ONE 12(5): e0176363. https://doi. tive disease of autosomal recessive inheritance. To date there is no biomarker for ACO2 org/10.1371/journal.pone.0176363 deficiency and diagnosis relies on genetic analysis. Here we report global metabolic profiling Editor: Petras Dzeja, Mayo Clinic Rochester, in eight patients with ACO2 deficiency. -

Aconitase: to Be Or Not to Be Inside Plant Glyoxysomes, That Is the Question
biology Review Aconitase: To Be or not to Be Inside Plant Glyoxysomes, That Is the Question Luigi De Bellis 1,* , Andrea Luvisi 1 and Amedeo Alpi 2 1 Department of Biological and Environmental Sciences and Technologies, University of Salento, Via Prov. le Monteroni, I-73100 Lecce, Italy; [email protected] 2 Approaching Research Educational Activities (A.R.E.A.) Foundation, I-56126 Pisa, Italy; [email protected] * Correspondence: [email protected] Received: 10 June 2020; Accepted: 10 July 2020; Published: 12 July 2020 Abstract: After the discovery in 1967 of plant glyoxysomes, aconitase, one the five enzymes involved in the glyoxylate cycle, was thought to be present in the organelles, and although this was found not to be the case around 25 years ago, it is still suggested in some textbooks and recent scientific articles. Genetic research (including the study of mutants and transcriptomic analysis) is becoming increasingly important in plant biology, so metabolic pathways must be presented correctly to avoid misinterpretation and the dissemination of bad science. The focus of our study is therefore aconitase, from its first localization inside the glyoxysomes to its relocation. We also examine data concerning the role of the enzyme malate dehydrogenase in the glyoxylate cycle and data of the expression of aconitase genes in Arabidopsis and other selected higher plants. We then propose a new model concerning the interaction between glyoxysomes, mitochondria and cytosol in cotyledons or endosperm during the germination of oil-rich seeds. Keywords: aconitase; malate dehydrogenase; glyoxylate cycle; glyoxysomes; peroxisomes; β-oxidation; gluconeogenesis 1. Introduction Glyoxysomes are specialized types of plant peroxisomes containing glyoxylate cycle enzymes, which participate in the conversion of lipids to sugar during the early stages of germination in oilseeds. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -

ACO2 Mutations: a Novel Phenotype Associating Severe Optic Atrophy and Spastic Paraplegia
CLINICAL/SCIENTIFIC NOTES OPEN ACCESS ACO2 mutations: A novel phenotype associating severe optic atrophy and spastic paraplegia Cecilia Marelli, MD, PhD, Christian Hamel, MD, PhD,† Melanie Quiles, Tech, Bertrand Carlander, MD, Correspondence Lise Larrieu, Tech, Cecile Delettre, PhD, Emmanuelle Sarzi, PhD, Dominique Chretien, PhD, Pierre Rustin, PhD, Dr. Guissart [email protected] Michel Koenig, MD, PhD, and Claire Guissart, PharmD, PhD Neurol Genet 2018;4:e225. doi:10.1212/NXG.0000000000000225 Aconitase 2 (ACO2) encodes the mitochondrial aconitase (ACO2), an enzyme catalyzing interconversion of citrate into isocitrate in the Krebs cycle. ACO2 mutations have been initially associated with infantile cerebellar-retinal degeneration combining optic atrophy, retinal – degeneration, severe encephalopathy, epilepsy, and cerebellar ataxia1 3; subsequently, ACO2 mutations have also been associated with milder presentations including isolated optic atrophy2 or cerebellar ataxia without optic atrophy.4 We report here a patient presenting with a novel ACO2 phenotype associating optic atrophy with spastic paraplegia. Methods Mini-exome sequencing by exon capture (TruSight One Sequencing Panel kit—Montpellier NGS platform) was performed in a patient with familial syndromic optic atrophy. Written informed consent was obtained. Aconitase enzymatic activity was measured as previously described.5 Case report A now 56-year-old white lady issued from a nonconsanguineous union presented because of severe optic atrophy and spastic paraplegia. Her symptoms had been slowly progressive since infancy and associated with delayed motor (walking at about 3 years) and mental development. An ophthalmologist (C.H.) first examined her at the age of 38 years because of her visual problems. She came to the neurologic clinic at the age of 49 years because of aggravation of the spastic paraplegia, requiring a walking aid.