Psykisk Utviklingshemming
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Centronuclear Myopathies Under Attack: a Plethora of Therapeutic Targets Hichem Tasfaout, Belinda Cowling, Jocelyn Laporte
CORE Metadata, citation and similar papers at core.ac.uk Provided by Archive Ouverte en Sciences de l'Information et de la Communication Centronuclear myopathies under attack: A plethora of therapeutic targets Hichem Tasfaout, Belinda Cowling, Jocelyn Laporte To cite this version: Hichem Tasfaout, Belinda Cowling, Jocelyn Laporte. Centronuclear myopathies under attack: A plethora of therapeutic targets. Journal of Neuromuscular Diseases, IOS Press, 2018, 5, pp.387 - 406. 10.3233/JND-180309. hal-02438924 HAL Id: hal-02438924 https://hal.archives-ouvertes.fr/hal-02438924 Submitted on 14 Jan 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Journal of Neuromuscular Diseases 5 (2018) 387–406 387 DOI 10.3233/JND-180309 IOS Press Review Centronuclear myopathies under attack: A plethora of therapeutic targets Hichem Tasfaouta,b,c,d, Belinda S. Cowlinga,b,c,d,1 and Jocelyn Laportea,b,c,d,1,∗ aDepartment of Translational Medicine and Neurogenetics, Institut de G´en´etique et de Biologie Mol´eculaire et Cellulaire (IGBMC), Illkirch, France bInstitut National de la Sant´eetdelaRechercheM´edicale (INSERM), U1258, Illkirch, France cCentre National de la Recherche Scientifique (CNRS), UMR7104, Illkirch, France dUniversit´e de Strasbourg, Illkirch, France Abstract. -

Affected Female Carriers of MTM1 Mutations Display a Wide Spectrum
Acta Neuropathol DOI 10.1007/s00401-017-1748-0 ORIGINAL PAPER Afected female carriers of MTM1 mutations display a wide spectrum of clinical and pathological involvement: delineating diagnostic clues Valérie Biancalana1,2,3,4,5 · Sophie Scheidecker1 · Marguerite Miguet1 · Annie Laquerrière6 · Norma B. Romero7,8 · Tanya Stojkovic8 · Osorio Abath Neto9 · Sandra Mercier10,11,12 · Nicol Voermans13 · Laura Tanner14 · Curtis Rogers15 · Elisabeth Ollagnon‑Roman16 · Helen Roper17 · Célia Boutte18 · Shay Ben‑Shachar19 · Xavière Lornage2,3,4,5 · Nasim Vasli2,3,4,5 · Elise Schaefer20 · Pascal Laforet21 · Jean Pouget22 · Alexandre Moerman23 · Laurent Pasquier24 · Pascale Marcorelle25,26 · Armelle Magot12 · Benno Küsters27 · Nathalie Streichenberger28 · Christine Tranchant29 · Nicolas Dondaine1 · Raphael Schneider2,3,4,5,30 · Claire Gasnier1 · Nadège Calmels1 · Valérie Kremer31 · Karine Nguyen32 · Julie Perrier12 · Erik Jan Kamsteeg33 · Pierre Carlier34 · Robert‑Yves Carlier35 · Julie Thompson30 · Anne Boland36 · Jean‑François Deleuze36 · Michel Fardeau7,8 · Edmar Zanoteli9 · Bruno Eymard21 · Jocelyn Laporte2,3,4,5 Received: 9 May 2017 / Revised: 24 June 2017 / Accepted: 2 July 2017 © Springer-Verlag GmbH Germany 2017 Abstract X-linked myotubular myopathy (XLMTM), a females and to delineate diagnostic clues, we character- severe congenital myopathy, is caused by mutations in the ized 17 new unrelated afected females and performed a MTM1 gene located on the X chromosome. A majority of detailed comparison with previously reported cases at the afected males die in the early postnatal period, whereas clinical, muscle imaging, histological, ultrastructural and female carriers are believed to be usually asymptomatic. molecular levels. Taken together, the analysis of this large Nevertheless, several afected females have been reported. cohort of 43 cases highlights a wide spectrum of clini- To assess the phenotypic and pathological spectra of carrier cal severity ranging from severe neonatal and generalized weakness, similar to XLMTM male, to milder adult forms. -

PTEN Mutations the PTEN Hamartoma Tumor Synd
Updated December 2019 (NCCN v1.2020) Cowden Syndrome/PTEN Hamartoma Tumor Syndrome: PTEN Mutations The PTEN Hamartoma Tumor Syndrome (PHTS) is a spectrum of highly variable conditions with overlapping features. This spectrum includes Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), and PTEN-related autism spectrum disorder.1-3 The term PHTS describes any individual with a germline pathogenic PTEN mutation, regardless of their clinical presentation.4 PHTS is a multisystem syndrome primarily characterized by noncancerous (benign), tumor-like growths called hamartomas that can develop throughout the body. There is also an increased risk of adult-onset cancers.5 Cancer Risks and General Management Recommendations PTEN Mutation General Surveillance/Management Recommendations9 Carrier Cancer Population Risks2,4-8 Lifetime Cancer Risks Female Breast: 12.4% Surveillance Primary: 33-60% Breast awareness, including periodic, consistent breast self exams, Second Primary: starting at age 18 years 29% within 10 Clinical breast exam every 6-12 months starting at age 25 years, or 5- years10 10 years before the earliest breast cancer diagnosis in the family (whichever comes first) Annual mammogram with consideration of tomosynthesis and breast MRI with contrast at age 30-35 years, or 5-10 years before the earliest breast cancer diagnosis in the family (whichever comes first) Age >75 years: Management should be considered on an individual basis Surgery Discuss option of risk-reducing mastectomy, including degree of protection, reconstruction -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Circle Applicable Codes
IDENTIFYING INFORMATION (please print legibly) Individual’s Name: DOB: Last 4 Digits of Social Security #: CIRCLE APPLICABLE CODES ICD-10 ICD-10 ICD-9 DIAGNOSTIC ICD-9 DIAGNOSTIC PRIMARY ICD-9 CODES CODE CODE PRIMARY ICD-9 CODES CODE CODE Abetalipoproteinemia 272.5 E78.6 Hallervorden-Spatz Syndrome 333.0 G23.0 Acrocephalosyndactyly (Apert’s Syndrome) 755.55 Q87.0 Head Injury, unspecified – Age of onset: 959.01 S09.90XA Adrenaleukodystrophy 277.86 E71.529 Hemiplegia, unspecified 342.9 G81.90 Arginase Deficiency 270.6 E72.21 Holoprosencephaly 742.2 Q04.2 Agenesis of the Corpus Callosum 742.2 Q04.3 Homocystinuria 270.4 E72.11 Agenesis of Septum Pellucidum 742.2 Q04.3 Huntington’s Chorea 333.4 G10 Argyria/Pachygyria/Microgyria 742.2 Q04.3 Hurler’s Syndrome 277.5 E76.01 or 758.33 Aicardi Syndrome 333 G23.8 Hyperammonemia Syndrome 270.6 E72.4 Alcohol Embryo and Fetopathy 760.71 F84.5 I-Cell Disease 272.2 E77.0 Anencephaly 655.0 Q00.0 Idiopathic Torsion Dystonia 333.6 G24.1 Angelman Syndrome 759.89 Q93.5 Incontinentia Pigmenti 757.33 Q82.3 Asperger Syndrome 299.8 F84.5 Infantile Cerebral Palsy, unspecified 343.9 G80.9 Ataxia-Telangiectasia 334.8 G11.3 Intractable Seizure Disorder 345.1 G40.309 Autistic Disorder (Childhood Autism, Infantile 299.0 F84.0 Klinefelter’s Syndrome 758.7 Q98.4 Psychosis, Kanner’s Syndrome) Biotinidase Deficiency 277.6 D84.1 Krabbe Disease 333.0 E75.23 Canavan Disease 330.0 E75.29 Kugelberg-Welander Disease 335.11 G12.1 Carpenter Syndrome 759.89 Q87.0 Larsen’s Syndrome 755.8 Q74.8 Cerebral Palsy, unspecified 343.69 G80.9 -

2018 Etiologies by Frequencies
2018 Etiologies in Order of Frequency by Category Hereditary Syndromes and Disorders Count CHARGE Syndrome 958 Down syndrome (Trisomy 21 syndrome) 308 Usher I syndrome 252 Stickler syndrome 130 Dandy Walker syndrome 119 Cornelia de Lange 102 Goldenhar syndrome 98 Usher II syndrome 83 Wolf-Hirschhorn syndrome (Trisomy 4p) 68 Trisomy 13 (Trisomy 13-15, Patau syndrome) 60 Pierre-Robin syndrome 57 Moebius syndrome 55 Trisomy 18 (Edwards syndrome) 52 Norrie disease 38 Leber congenital amaurosis 35 Chromosome 18, Ring 18 31 Aicardi syndrome 29 Alstrom syndrome 27 Pfieffer syndrome 27 Treacher Collins syndrome 27 Waardenburg syndrome 27 Marshall syndrome 25 Refsum syndrome 21 Cri du chat syndrome (Chromosome 5p- synd) 16 Bardet-Biedl syndrome (Laurence Moon-Biedl) 15 Hurler syndrome (MPS I-H) 15 Crouzon syndrome (Craniofacial Dysotosis) 13 NF1 - Neurofibromatosis (von Recklinghausen dis) 13 Kniest Dysplasia 12 Turner syndrome 11 Usher III syndrome 10 Cockayne syndrome 9 Apert syndrome/Acrocephalosyndactyly, Type 1 8 Leigh Disease 8 Alport syndrome 6 Monosomy 10p 6 NF2 - Bilateral Acoustic Neurofibromatosis 6 Batten disease 5 Kearns-Sayre syndrome 5 Klippel-Feil sequence 5 Hereditary Syndromes and Disorders Count Prader-Willi 5 Sturge-Weber syndrome 5 Marfan syndrome 3 Hand-Schuller-Christian (Histiocytosis X) 2 Hunter Syndrome (MPS II) 2 Maroteaux-Lamy syndrome (MPS VI) 2 Morquio syndrome (MPS IV-B) 2 Optico-Cochleo-Dentate Degeneration 2 Smith-Lemli-Opitz (SLO) syndrome 2 Wildervanck syndrome 2 Herpes-Zoster (or Hunt) 1 Vogt-Koyanagi-Harada -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -
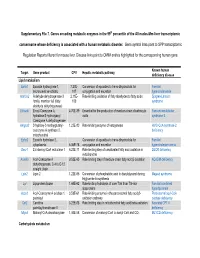
Supplementary File 7. Genes Encoding Metabolic Enzymes in The
Supplementary File 7. Genes encoding metabolic enzymes in the 99th percentile of the All nodes-Mm-liver transcriptomic consensome whose deficiency is associated with a human metabolic disorder. Gene symbol links point to SPP transcriptomic Regulation Reports filtered for mouse liver. Disease links point to OMIM entries highlighted for the corresponding human gene. Known human Target Gene product CPV Hepatic metabolic pathway deficiency disease Lipid metabolism Ephx1 Epoxide hydroxylase 1, 7.27E- Conversion of epoxides to trans-dihydrodiols for Familial microsomal xenobiotic 107 conjugation and excretion hypercholanemia Aldh3a2 Aldehyde dehydrogenase 3 2.11E- Rate-limiting oxidation of fatty aldehydes to fatty acids Sjogren-Larsson family, member A2 (fatty 103 syndrome aldehyde dehydrogenase) Ehhadh Enoyl-Coenzyme A, 4.70E-89 Essential for the production of medium-chain dicarboxylic Fanconi renotubular hydratase/3-hydroxyacyl acids syndrome 3 Coenzyme A dehydrogenase Hmgcs2 3-hydroxy-3-methylglutaryl- 1.23E-80 Rate-limiting enzyme of ketogenesis HMG-CoA synthase-2 coenzyme A synthase 2, deficiency mitochondrial Ephx2 Epoxide hydrolase 2, Conversion of epoxides to trans-dihydrodiols for Familial cytoplasmic 6.06E-78 conjugation and excretion hypercholesterolemia Decr1 2,4-dienoyl-CoA reductase 1 6.23E-71 Rate-limiting step of unsaturated fatty acid oxidation in DECR deficiency mitochondria Acadm Acyl-Coenzyme A 9.52E-65 Rate limiting step of medium-chain fatty acid β-oxidation ACADM deficiency dehydrogenase, C-4 to C-12 straight chain Lpin2 -

Diseases of the Digestive System (KOO-K93)
CHAPTER XI Diseases of the digestive system (KOO-K93) Diseases of oral cavity, salivary glands and jaws (KOO-K14) lijell Diseases of pulp and periapical tissues 1m Dentofacial anomalies [including malocclusion] Excludes: hemifacial atrophy or hypertrophy (Q67.4) K07 .0 Major anomalies of jaw size Hyperplasia, hypoplasia: • mandibular • maxillary Macrognathism (mandibular)(maxillary) Micrognathism (mandibular)( maxillary) Excludes: acromegaly (E22.0) Robin's syndrome (087.07) K07 .1 Anomalies of jaw-cranial base relationship Asymmetry of jaw Prognathism (mandibular)( maxillary) Retrognathism (mandibular)(maxillary) K07.2 Anomalies of dental arch relationship Cross bite (anterior)(posterior) Dis to-occlusion Mesio-occlusion Midline deviation of dental arch Openbite (anterior )(posterior) Overbite (excessive): • deep • horizontal • vertical Overjet Posterior lingual occlusion of mandibular teeth 289 ICO-N A K07.3 Anomalies of tooth position Crowding Diastema Displacement of tooth or teeth Rotation Spacing, abnormal Transposition Impacted or embedded teeth with abnormal position of such teeth or adjacent teeth K07.4 Malocclusion, unspecified K07.5 Dentofacial functional abnormalities Abnormal jaw closure Malocclusion due to: • abnormal swallowing • mouth breathing • tongue, lip or finger habits K07.6 Temporomandibular joint disorders Costen's complex or syndrome Derangement of temporomandibular joint Snapping jaw Temporomandibular joint-pain-dysfunction syndrome Excludes: current temporomandibular joint: • dislocation (S03.0) • strain (S03.4) K07.8 Other dentofacial anomalies K07.9 Dentofacial anomaly, unspecified 1m Stomatitis and related lesions K12.0 Recurrent oral aphthae Aphthous stomatitis (major)(minor) Bednar's aphthae Periadenitis mucosa necrotica recurrens Recurrent aphthous ulcer Stomatitis herpetiformis 290 DISEASES OF THE DIGESTIVE SYSTEM Diseases of oesophagus, stomach and duodenum (K20-K31) Ill Oesophagitis Abscess of oesophagus Oesophagitis: • NOS • chemical • peptic Use additional external cause code (Chapter XX), if desired, to identify cause. -

Prevalence and Incidence of Rare Diseases: Bibliographic Data
Number 1 | January 2019 Prevalence and incidence of rare diseases: Bibliographic data Prevalence, incidence or number of published cases listed by diseases (in alphabetical order) www.orpha.net www.orphadata.org If a range of national data is available, the average is Methodology calculated to estimate the worldwide or European prevalence or incidence. When a range of data sources is available, the most Orphanet carries out a systematic survey of literature in recent data source that meets a certain number of quality order to estimate the prevalence and incidence of rare criteria is favoured (registries, meta-analyses, diseases. This study aims to collect new data regarding population-based studies, large cohorts studies). point prevalence, birth prevalence and incidence, and to update already published data according to new For congenital diseases, the prevalence is estimated, so scientific studies or other available data. that: Prevalence = birth prevalence x (patient life This data is presented in the following reports published expectancy/general population life expectancy). biannually: When only incidence data is documented, the prevalence is estimated when possible, so that : • Prevalence, incidence or number of published cases listed by diseases (in alphabetical order); Prevalence = incidence x disease mean duration. • Diseases listed by decreasing prevalence, incidence When neither prevalence nor incidence data is available, or number of published cases; which is the case for very rare diseases, the number of cases or families documented in the medical literature is Data collection provided. A number of different sources are used : Limitations of the study • Registries (RARECARE, EUROCAT, etc) ; The prevalence and incidence data presented in this report are only estimations and cannot be considered to • National/international health institutes and agencies be absolutely correct. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Complex Hereditary Spastic Paraplegia Associated with Episodic
Tozawa et al. Human Genome Variation (2021) 8:4 https://doi.org/10.1038/s41439-021-00136-y Human Genome Variation DATA REPORT Open Access Complex hereditary spastic paraplegia associated with episodic visual loss caused by ACO2 variants Takenori Tozawa1,2,AkiraNishimura3, Tamaki Ueno2,4, Akane Shikata5, Yoshihiro Taura1,TakeshiYoshida 6, Naoko Nakagawa7, Takahito Wada 7, Shinji Kosugi7, Tomoko Uehara8, Toshiki Takenouchi 9, Kenjiro Kosaki8 and Tomohiro Chiyonobu1 Abstract Most patients with homozygous or compound heterozygous pathogenic ACO2 variants present with muscular hypotonia features, namely, infantile cerebellar-retinal degeneration. Recently, two studies reported rare familial cases of ACO2 variants presenting as complex hereditary spastic paraplegia (HSP) with broad clinical spectra. Here, we report the case of a 20-year-old Japanese woman with complex HSP caused by compound heterozygous ACO2 variants, revealing a new phenotype of episodic visual loss during febrile illness. The ACO2 gene on chromosome 22 encodes the aco- variants in the ACO2 gene presenting as complex her- nitase 2 (ACO2) protein in the mitochondrial matrix; editary spastic paraplegia (HSP) with a new phenotype of ACO2 catalyzes the stereospecific isomerization of citrate episodic visual loss after every febrile infection and pro- to isocitrate in the tricarboxylic acid (TCA) cycle1. gressive optic atrophy. This is the third familial report and ACO2 fi fi 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; Pathogenic variants were rst reported in eight the rst Asian patient with complex HSP caused by individuals from two Arab families, and they had infantile pathogenic ACO2 variants. cerebellar-retinal degeneration (ICRD, OMIM#614559)2. The proband was born to nonconsanguineous healthy Subsequently, ~20 cases of pathogenic homozygous or parents at 38 weeks gestational age after unremarkable compound heterozygous ACO2 variants have been delivery.