Foot Malformation with Or Without L
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Mechanisms of Pitx1 Action in Murine Hindlimb Development
1 Genetic mechanisms of Pitx1 action in murine hindlimb development Stephen Nemec, Division of Experimental Medicine, McGill University, Montreal August 2017 A thesis submitted to McGill University in partial fulfillment of the degree of PhD © Stephen Nemec 2017 2 Table of Contents Contents Page Abstract 4 Acknowledgements 8 Abbreviations 9 Preface – Contribution to knowledge 10 Contribution of authors 11 Introduction 13 Figures 1 and 2: Basics of limb anatomy and development 13 Evolutionary origins of the limb 14 Chick embryology and the early study of the limb 16 Molecular limb development 21 Hox genes – Engines of limb development 25 The genetics of forelimb vs. hindlimb development 30 Pitx1: major regulator of HL-specific pattern 30 Tbx4 and Tbx5 – limb-type-specific Tbox paralogs 35 Tbx4, Tbx5 and developmental anomalies in humans 41 Pitx1 Tbx4 42 Purpose and Aims 45 Pitx1 directly modulates the core limb development 46 program to implement hindlimb identity Contributions 47 Abstract 48 Introduction 49 Results 51 Discussion 60 Materials and Methods 65 Figure Legends 69 Figures 74 Interlude A – From Sox9 to signaling 89 Shh signaling influences the 91 phenotype of Pitx1-/- hindlimbs Contributions 92 Abstract 93 Introduction 94 Results 96 Discussion 98 Materials and Methods 100 Figure Legends 102 Figures 104 Interlude B – Regulatory complexity and developmental constraints 110 3 Table of Contents (continued) Contents Page Regulatory integration of Hox factor action with 111 Tbox factors in limb development Contributions 112 Abstract 113 Introduction 114 Results 116 Discussion 124 Materials and Methods 128 Figure Legends 134 Figures 141 Discussion 152 Evolutionary constraints determine the 152 developmental roles of limb-type-specific genes Future Directions 156 References 159 4 Abstract In tetrapods, the forelimbs (FL) and hindlimbs (HL) emerge from the flank of the developing embryo as buds of mesenchyme sheathed in ectoderm. -

<Abstract Centered> an ABSTRACT of the THESIS OF
AN ABSTRACT OF THE DISSERTATION OF Michael Austin Garland for the degree of Doctor of Philosophy in Toxicology presented on June 14, 2019. Title: Transcriptomic Approaches for Discovering Regenerative and Developmental Regulatory Networks in Zebrafish Abstract approved: _____________________________________________________________________ Robert L. Tanguay Zebrafish are capable of fully regenerating organs and tissue such as their caudal fin, which is similar to a human regrowing an arm or a leg. In contrast, most mammals including humans have a greatly reduced capacity for wound healing. The ability of zebrafish to undergo this regenerative process, called epimorphic regeneration, hinges on the capacity to form a blastema at the wound site. The blastema quickly recapitulates the developmental processes involved in complex tissue formation to restore lost or damaged tissue. One key mechanism for inducing blastema formation is global repression of genes involved in tissue differentiation and maintenance. Induction of repressive factors, such as microRNAs (miRNAs), are involved in reprogramming cells during epimorphic regeneration. The upstream mechanism by which zebrafish undergo epimorphic regeneration remains elusive. Furthermore, while focus is shifting toward regulatory RNAs such as miRNAs, the full complement of their repressive activities is unknown. We took a transcriptomics approach to investigating epimorphic regeneration and fin development. Parallel sequencing of total RNA and small RNA samples was performed on regenerating fin tissue at 1 day post-amputation (dpa). Most miRNAs had increased expression, consistent with global repression of genes involved in cell specialization during de-differentiation. We identified predicted interactions between miRNAs and genes involved in transcriptional regulation, chromatin modification, and developmental signaling. miR-146a and miR-146b are anti- inflammatory miRNAs that were predicted to target eya4, which is involved in chromatin remodeling and innate immunity. -

Genome-Wide Screening Identifies Genes and Biological Processes
Louisiana State University LSU Digital Commons LSU Doctoral Dissertations Graduate School 10-12-2018 Genome-Wide Screening Identifies Genes and Biological Processes Implicated in Chemoresistance and Oncogene-Induced Apoptosis Tengyu Ko Louisiana State University and Agricultural and Mechanical College, [email protected] Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_dissertations Part of the Cancer Biology Commons, Cell Biology Commons, and the Genomics Commons Recommended Citation Ko, Tengyu, "Genome-Wide Screening Identifies Genes and Biological Processes Implicated in Chemoresistance and Oncogene- Induced Apoptosis" (2018). LSU Doctoral Dissertations. 4715. https://digitalcommons.lsu.edu/gradschool_dissertations/4715 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Doctoral Dissertations by an authorized graduate school editor of LSU Digital Commons. For more information, please [email protected]. GENOME-WIDE SCREENING IDENTIFIES GENES AND BIOLOGICAL PROCESSES IMPLICATED IN CHEMORESISTANCE AND ONCOGENE- INDUCED APOPTOSIS A Dissertation Submitted to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical and Veterinary Medical Sciences through the Department of Comparative Biomedical Sciences by Tengyu Ko B.S., University of California, Santa Barbara 2010 December 2018 ACKNOWLEDGEMENTS I would like to express my sincerest gratitude to my major supervisor Dr. Shisheng Li for giving me the opportunity to join his team and the freedom to pursue projects. I appreciate all of his thoughts and efforts. Truly, none of these findings would be possible without his supervisions, supports, insightful discussions, and patience. -

Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal Through Mirna-1247-3P in Human Cervical Cancer Cells
Article Volume 11, Issue 1, 2021, 7943 - 7972 https://doi.org/10.33263/BRIAC111.79437972 Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal through miRNA-1247-3p in Human Cervical Cancer Cells Meng Luo 1 , Dian Fan 2 , Yinan Xiao 2 , Hao Zeng 2 , Dingyue Zhang 2 , Yunuo Zhao 2 , Xuelei Ma 1,* 1 Department of Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (L.M.); 2 West China Medical School, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (F.D.); [email protected] (X.Y.N.); [email protected] (Z.H.); [email protected] (Z.D.Y.); [email protected] (Z.Y.N.); * Correspondence: [email protected]; Scopus Author ID 55523405000 Received: 11.06.2020; Revised: 3.07.2020; Accepted: 5.07.2020; Published: 9.07.2020 Abstract: The prognosis of cervical cancer remains poor. Sulforaphane, an active ingredient from cruciferous plants, has been identified as a potential anticancer agent in various cancers. However, there is little information about its effect on cervical cancer. Here, we conducted a present study to uncover the effect and the potential mechanisms of sulforaphane on cervical cancer. HeLa cells were treated with sulforaphane, and cell proliferation and apoptosis were assessed by Cell Counting Kit-8, Western blot, flow cytometry, and immunofluorescence. Then, next-generation sequencing (NGS) and bioinformatics tools were used to analyze mRNA-seq, miRNA-seq, and potential pathways. Finally, qRT-PCR, Cell Counting Kit-8, flow cytometry, small RNAs analysis, and Western blot were performed to evaluate the biological function of miR1247-3p and MAPK pathway in HeLa cell lines. -

The Changing Chromatome As a Driver of Disease: a Panoramic View from Different Methodologies
The changing chromatome as a driver of disease: A panoramic view from different methodologies Isabel Espejo1, Luciano Di Croce,1,2,3 and Sergi Aranda1 1. Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain 2. Universitat Pompeu Fabra (UPF), Barcelona, Spain 3. ICREA, Pg. Lluis Companys 23, Barcelona 08010, Spain *Corresponding authors: Luciano Di Croce ([email protected]) Sergi Aranda ([email protected]) 1 GRAPHICAL ABSTRACT Chromatin-bound proteins regulate gene expression, replicate and repair DNA, and transmit epigenetic information. Several human diseases are highly influenced by alterations in the chromatin- bound proteome. Thus, biochemical approaches for the systematic characterization of the chromatome could contribute to identifying new regulators of cellular functionality, including those that are relevant to human disorders. 2 SUMMARY Chromatin-bound proteins underlie several fundamental cellular functions, such as control of gene expression and the faithful transmission of genetic and epigenetic information. Components of the chromatin proteome (the “chromatome”) are essential in human life, and mutations in chromatin-bound proteins are frequently drivers of human diseases, such as cancer. Proteomic characterization of chromatin and de novo identification of chromatin interactors could thus reveal important and perhaps unexpected players implicated in human physiology and disease. Recently, intensive research efforts have focused on developing strategies to characterize the chromatome composition. In this review, we provide an overview of the dynamic composition of the chromatome, highlight the importance of its alterations as a driving force in human disease (and particularly in cancer), and discuss the different approaches to systematically characterize the chromatin-bound proteome in a global manner. -
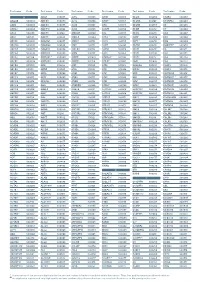
Aagab S00002 Aars S00003 Aars2 S00004 Aass S02483
Test name Code Test name Code Test name Code Test name Code Test name Code Test name Code A ADAR S00053 ALPL S00105 ARSB S00153 BCL10 S02266 C5AR2 S00263 AAGAB S00002 ADCK3 S00054 ALS2 S00106 ARSE * S00154 BCL11A S02167 C5ORF42 S00264 AARS S00003 ADCK4 S00055 ALX3 S00107 ARX S00155 BCL11B S02358 C6 S00265 AARS2 S00004 ADCY10 S02094 ALX4 S00108 ASAH1 S00156 BCOR S00212 C7 S00266 AASS S02483 ADCY3 S02184 AMACR S00109 ASL S00157 BCS1L S00213 C8A S00267 ABAT S02191 ADCY5 S02226 AMELX S02289 ASNS * S02508 BDNF S02509 C8B S00268 ABCA1 S00005 ADGRG1 S00057 AMER1 S00110 ASPA S00158 BDP1 * S00214 C8G S00269 ABCA12 S00006 ADGRG6 S02548 AMH S00111 ASPH S02425 BEAN1 S00215 C8ORF37 S00270 ABCA3 S00007 ADGRV1 S00058 AMHR2 S00112 ASPM S00159 BEST1 S00216 C9 S00271 ABCA4 S00008 ADIPOQ S00059 AMN S00113 ASS1 S00160 BFSP1 S02280 CA2 S00272 ABCA7 S02106 ADIPOR1 * S00060 AMPD1 S02670 ATAD3A * S02196 BFSP2 S00217 CA4 S02303 ABCB11 S00009 ADIPOR2 S00061 AMPD2 S02128 ATCAY S00162 BGN S02633 CA8 S00273 ABCB4 S00010 ADK S02595 AMT S00114 ATF6 S00163 BHLHA9 S00218 CABP2 S00274 ABCB6 S00011 ADNP S02320 ANG S00115 ATIC S02458 BICD2 S00220 CABP4 S00275 ABCB7 S00012 ADSL S00062 ANK1 S00116 ATL1 S00164 BIN1 S00221 CACNA1A S00276 ABCC2 S00013 AFF2 S00063 ANK2 S00117 ATL3 S00165 BLK S00222 CACNA1C * S00277 ABCC6 * S00014 AFG3L2 * S00064 ANKH S00118 ATM S00166 BLM S00223 CACNA1D S00278 ABCC8 S00015 AGA S00065 ANKRD11 * S02140 ATOH7 S02390 BLNK S02281 CACNA1F S00279 ABCC9 S00016 AGBL5 S02452 ANKS6 S00121 ATP13A2 S00168 BLOC1S3 S00224 CACNA1H S00280 ABCD1 * S00017 AGK * -

Predict AID Targeting in Non-Ig Genes Multiple Transcription Factor
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 is online at: average * The Journal of Immunology published online 20 March 2013 from submission to initial decision 4 weeks from acceptance to publication Multiple Transcription Factor Binding Sites Predict AID Targeting in Non-Ig Genes Jamie L. Duke, Man Liu, Gur Yaari, Ashraf M. Khalil, Mary M. Tomayko, Mark J. Shlomchik, David G. Schatz and Steven H. Kleinstein J Immunol http://www.jimmunol.org/content/early/2013/03/20/jimmun ol.1202547 Submit online. Every submission reviewed by practicing scientists ? is published twice each month by http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://www.jimmunol.org/content/suppl/2013/03/20/jimmunol.120254 7.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. Published March 20, 2013, doi:10.4049/jimmunol.1202547 The Journal of Immunology Multiple Transcription Factor Binding Sites Predict AID Targeting in Non-Ig Genes Jamie L. Duke,* Man Liu,†,1 Gur Yaari,‡ Ashraf M. Khalil,x Mary M. Tomayko,{ Mark J. Shlomchik,†,x David G. -

Genetic Regulatory Pathways of Split Hand-Foot Malformation
Received: 28 July 2018 Accepted: 7 August 2018 DOI: 10.1111/cge.13434 REVIEW Genetic regulatory pathways of split-hand/foot malformation Piranit N. Kantaputra1,2,3 | Bruce M. Carlson4 1Center of Excellence in Medical Genetics Research, Chiang Mai University, Chiang Mai, Split-hand/foot malformation (SHFM) is caused by mutations in TP63, DLX5, DLX6, FGF8, Thailand FGFR1, WNT10B, and BHLHA9. The clinical features of SHFM caused by mutations of these 2Division of Pediatric Dentistry, Department genes are not distinguishable. This implies that in normal situations these SHFM-associated of Orthodontics and Pediatric Dentistry, genes share an underlying regulatory pathway that is involved in the development of the central Faculty of Dentistry, Chiang Mai University, Chiang Mai, Thailand parts of the hands and feet. The mutations in SHFM-related genes lead to dysregulation of Fgf8 3Dentaland Clinic, Chiang Mai, Thailand in the central portion of the apical ectodermal ridge (AER) and subsequently lead to misexpres- 4Department of Anatomy and Cell Biology, sion of a number of downstream target genes, failure of stratification of the AER, and thus University of Michigan, Ann Arbor, Michigan SHFM. Syndactyly of the remaining digits is most likely the effects of dysregulation of Fgf-Bmp- Correspondence Msx signaling on apoptotic cell death. Loss of digit identity in SHFM is hypothesized to be the Piranit N. Kantaputra, Division of Pediatric effects of misexpression of HOX genes, abnormal SHH gradient, or the loss of balance between Dentistry, Department of Orthodontics and GLI3A and GLI3R. Disruption of canonical and non-canonical Wnt signaling is involved in the Pediatric Dentistry, Faculty of Dentistry, Chiang Mai University, Dentaland Clinic, pathogenesis of SHFM. -

Macaque Gene Symbols Were Mapped to Human Gene Symbols on June 2011
This document is a guide for cross referencing human gene symbols to macaque gene symbols. Note: macaque gene symbols were mapped to human gene symbols on June 2011. Macaque transcript accession number was used to obtain macaque transcript sequence. Then, the macaque transcript sequence was blasted against human refseq. Highest scoring blast match with a minimum bit score cutoff of 200 was selected. Resulting human gene symbol was then mapped to the corresponding macaque gene symbol. An Excel file of this document is available for downloading at http://download.alleninstitute.org:80/nhp/ Macaque Macaque Human Human Gene Symbol Entrez Id Gene Symbol Entrez ID A1BG 712737 A1BG 1 A1CF 703806 A1CF 29974 A1CF 703806 A1CF 29974 LOC708209 708209 A2BP1 54715 LOC713147 713147 A2BP1 54715 LOC706006 706006 A2LD1 87769 LOC706006 706006 A2LD1 87769 A2M 716834 A2M 2 LOC722289 722289 A2M 2 A2ML1 716616 A2ML1 144568 A4GALT 710998 A4GALT 53947 A4GALT 710998 A4GALT 53947 A4GALT 710998 A4GALT 53947 A4GNT 716512 A4GNT 51146 LOC699771 699771 AAAS 8086 LOC719095 719095 AAAS 8086 AACS 707015 AACS 65985 LOC100427856 100427856 AACSL 729522 AADAC 709031 AADAC 13 AADACL2 709222 AADACL2 344752 AADACL2 709222 AADACL2 344752 LOC711944 711944 AADACL2 344752 LOC722778 722778 AADACL3 126767 AADACL4 715600 AADACL4 343066 AADAT 695264 AADAT 51166 LOC711436 711436 AAGAB 79719 LOC701067 701067 AAK1 22848 LOC100429994 100429994 AAK1 22848 LOC100430095 100430095 AAK1 22848 LOC100430344 100430344 AAK1 22848 AAMP 700763 AAMP 14 AANAT 706924 AANAT 15 AARS 709492 AARS 16 AARS2 -

Class a Basic Helix Loop Helix Protein 9 (BHLHA9) (NM 001164405) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC228166 Class A basic helix loop helix protein 9 (BHLHA9) (NM_001164405) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Class A basic helix loop helix protein 9 (BHLHA9) (NM_001164405) Human Tagged ORF Clone Tag: Myc-DDK Symbol: BHLHA9 Synonyms: BHLHF42; CCSPD Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC228166 representing NM_001164405 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGCTGCGGGGCGCGCCAGGACTAGGCCTCACGGCGCGGAAGGGGGCCGAGGACTCTGCGGAGGACTTGG GGGGCCCCTGCCCCGAGCCCGGGGGCGATTCGGGGGTGCTGGGGGCGAACGGCGCTTCCTGCAGCCGGGG CGAGGCGGAGGAGCCGGCGGGCAGGAGGCGCGCGCGGCCGGTGCGGTCCAAGGCGCGGCGCATGGCCGCC AACGTGCGGGAGCGCAAGCGCATCCTAGACTACAACGAGGCCTTCAACGCGCTGCGCCGGGCGCTGCGGC ACGACCTGGGCGGCAAGAGGCTCTCCAAGATCGCCACGCTGCGCAGGGCCATCCACCGCATCGCCGCGCT CTCCCTGGTCCTGCGCGCCAGCCCCGCGCCCCGCGGGCCCTGCGGACACCTGGAGTGCCACGGCCCGGCC GCGCGCGGGGACACCGGGGACACAGGCGCCAGCCCCCCGCCGCCTGCAGGGCCCAGCCTCGCGCGCCCAG ACGCCGCCCGCCCCTCGGTGCCGTCCGCGCCCCGCTGCGCCTCGTGCCCCCCGCACGCGCCCCTGGCACG GCCCAGTGCGGTGGCCGAGGGGCCGGGCCTAGCACAGGCCTCCGGGGGAAGCTGGCGCCGCTGTCCGGGG GCTTCCTCTGCCGGGCCGCCTCCCTGGCCGCGGGGCTACCTGCGATCCGCCCCCGGGATGGGCCATCCGC GCTCC ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT -

Table S1. Candidate Genes for the Phenotypes Polydactyly, Syndactyly and Polysyndactyly in Domestic Animals According to NCBI and OMIA
Table S1. Candidate genes for the phenotypes polydactyly, syndactyly and polysyndactyly in domestic animals according to NCBI and OMIA. The bovine orthologues genes were presented with their chromosomal position. Phenotype Gene Gene full name Species Gene ID Bovine gene BTA Position Syndactyly ADAMTS5 a disintegrin-like and Mus musculus ENSBTAG00000000648 ADAMTS5 1 8811792-8861602 metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 5 (aggrecanase-2) Polydactyly ARL13B ADP ribosylation factor like Homo sapiens ENSBTAG00000005155 ARL13B 1 37875395-37952674 GTPase 13B Polydactyly ARL6 ADP ribosylation factor like Homo sapiens ENSBTAG00000010091 ARL6 1 41640353-41679711 GTPase 6 Polydactyly Dzip1l DAZ interacting protein 1-like Mus musculus ENSBTAG00000015198 DZIP1L 1 132103024-132129289 Polydactyly Etv5 ets variant 5 Mus musculus ENSBTAG00000014915 ETV5 1 81725703-81784999 Polydactyly IFT80 intraflagellar transport 80 Homo sapiens ENSBTAG00000031572 IFT80 1 107962966-108058677 Polydactyly, PIK3CA phosphatidylinositol-4,5- Homo sapiens ENSBTAG00000009232 PIK3CA 1 88504290-88533061 syndactyly bisphosphate 3-kinase catalytic subunit alpha Polydactyly TCTEX1D2 Tctex1 domain containing 2 Homo sapiens ENSBTAG00000032684 TCTEX1D2 1 71489285-71516789 Polydactyly BBS5 Bardet-Biedl syndrome 5 Homo sapiens ENSBTAG00000010736 BBS5 2 26800661-26821659 Polydactyly CCDC28B coiled-coil domain containing Homo sapiens ENSBTAG00000015104 CCDC28B 2 122150084-122154154 28B Polydactyly, GLI2 GLI family zinc finger 2 Homo sapiens ENSBTAG00000011682 -

Dynamics and Function of Distal Regulatory Elements During Neurogenesis and Neuroplasticity
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Dynamics and Function of Distal Regulatory Elements during Neurogenesis and Neuroplasticity Sudhir Thakurela1,#, Sanjeeb Kumar Sahu1,#, Angela Garding1, Vijay K. Tiwari1,* 1. Institute of Molecular Biology (IMB), Mainz, Germany *Correspondence should be addressed to [email protected]. #These authors contributed equally to this work. Running title: Dynamics of distal regulatory landscape during neurogenesis and neuronal activity Key words: Gene regulation, epigenetic mechanisms, transcription factors, chromatin accessibility, neurogenesis, neuronal activity 1 Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press ABSTRACT Gene regulation in mammals involves a complex interplay between promoters and distal regulatory elements that function in concert to drive precise spatio-temporal gene expression programs. However, the dynamics of distal gene regulatory landscape and its function in the transcriptional reprogramming that underlies neurogenesis and neuronal activity remain largely unknown. Here, we performed a combinatorial analysis of genome-wide datasets for chromatin accessibility (FAIRE- seq) and the enhancer mark H3K27ac that reveal the highly dynamic nature of distal gene regulation during neurogenesis, which gets progressively restricted to distinct genomic regions as neurons acquire a post-mitotic, terminally differentiated state. We further find that the distal accessible and active regions serve as target sites for distinct transcription factors that function in a stage-specific manner to contribute to the transcriptional program underlying neuronal commitment and maturation. Mature neurons respond to a sustained activity of NMDA receptors by epigenetic reprogramming at a large number of distal regulatory regions as well as dramatic reorganization of super-enhancers.