In Silico Molecular Study of Tryptophan Bitterness
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

(12) United States Patent (10) Patent No.: US 9,347,934 B2 Shekdar Et Al
USOO9347934B2 (12) United States Patent (10) Patent No.: US 9,347,934 B2 Shekdar et al. (45) Date of Patent: May 24, 2016 (54) ASSAYS FOR IDENTIFYING COMPOUNDS 2008, OO38739 A1 2/2008 Li et al. THAT MODULATE BITTERTASTE 2008/0167286 A1* 7/2008 Gopalakrishnan et al. ........................ 514,21016 (71) Applicants: CHROMOCELL CORPORATION, 2010/01298.33 A1* 5/2010 Brune et al. ................. 435/721 North Brunswick, NJ (US); KRAFT FOODS GROUP BRANDS LLC, FOREIGN PATENT DOCUMENTS Northfield, IL (US) CN 1341632 A 3, 2002 CN 101583717 A 11, 2009 (72) Inventors: Kambiz Shekdar, New York, NY (US); CN 101828.111 A 9, 2010 Purvi Manoj Shah, Bridgewater, NJ WO WO-0038536 A2 7, 2000 WO WO-2004O29087 4/2004 (US); Joseph Gunnet, Flemington, NJ WO WO-2006053771 A2 5, 2006 (US); Jane V. Leland, Wilmette, IL WO WO-2007002026 A2 1/2007 (US); Peter H. Brown, Glenview, IL WO WO-2008057470 5, 2008 (US); Louise Slade, Morris Plains, NJ WO WO-2008119.195 A1 10, 2008 (US) WO WO-20081191.96 10, 2008 WO WO-20081191.97 10, 2008 (73) Assignees: Chromocell Corporation, North W WSi. A2 1929 Brunswick, NJ (US); Kraft Foods WO WO-2010O886.33 8, 2010 Group Brands LLC, Northfield, IL WO WO-2010O99983 A1 9, 2010 (US) WO WO-2013022947 2, 2013 (*) Notice: Subject to any disclaimer, the term of this OTHER PUBLICATIONS patent is extended or adjusted under 35 U.S.C. 154(b) by 0 days. Bachmanov et al., Taste Receptor Genes, 2007, 27:389-414.* Behrens et al., Structural Requirements for Bitter Taste Receptor (21) Appl. -

G Protein-Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. British Journal of Pharmacology (2015) 172, 5744–5869 THE CONCISE GUIDE TO PHARMACOLOGY 2015/16: G protein-coupled receptors Stephen PH Alexander1, Anthony P Davenport2, Eamonn Kelly3, Neil Marrion3, John A Peters4, Helen E Benson5, Elena Faccenda5, Adam J Pawson5, Joanna L Sharman5, Christopher Southan5, Jamie A Davies5 and CGTP Collaborators 1School of Biomedical Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK, 2Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK, 3School of Physiology and Pharmacology, University of Bristol, Bristol, BS8 1TD, UK, 4Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK, 5Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2015/16 provides concise overviews of the key properties of over 1750 human drug targets with their pharmacology, plus links to an open access knowledgebase of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. The full contents can be found at http://onlinelibrary.wiley.com/doi/ 10.1111/bph.13348/full. G protein-coupled receptors are one of the eight major pharmacological targets into which the Guide is divided, with the others being: ligand-gated ion channels, voltage-gated ion channels, other ion channels, nuclear hormone receptors, catalytic receptors, enzymes and transporters. These are presented with nomenclature guidance and summary information on the best available pharmacological tools, alongside key references and suggestions for further reading. -

G Protein‐Coupled Receptors
S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: G protein-coupled receptors. British Journal of Pharmacology (2019) 176, S21–S141 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors Stephen PH Alexander1 , Arthur Christopoulos2 , Anthony P Davenport3 , Eamonn Kelly4, Alistair Mathie5 , John A Peters6 , Emma L Veale5 ,JaneFArmstrong7 , Elena Faccenda7 ,SimonDHarding7 ,AdamJPawson7 , Joanna L Sharman7 , Christopher Southan7 , Jamie A Davies7 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Monash Institute of Pharmaceutical Sciences and Department of Pharmacology, Monash University, Parkville, Victoria 3052, Australia 3Clinical Pharmacology Unit, University of Cambridge, Cambridge, CB2 0QQ, UK 4School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 5Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 6Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 7Centre for Discovery Brain Sciences, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. Although the Concise Guide represents approximately 400 pages, the material presented is substantially reduced compared to information and links presented on the website. -

Genetic Variations in TAS2R3 and TAS2R4 Bitterness Receptors Modify
www.nature.com/scientificreports OPEN Genetic variations in TAS2R3 and TAS2R4 bitterness receptors modify papillary carcinoma risk and Received: 10 January 2018 Accepted: 25 September 2018 thyroid function in Korean females Published: xx xx xxxx Jeong-Hwa Choi 1,2, Jeonghee Lee 1, Sarah Yang1,3, Eun Kyung Lee4, Yul Hwangbo4 & Jeongseon Kim 1 Type 2 taste receptors (T2Rs, TAS2Rs) mediate bitterness perception and are involved in diverse defence mechanisms in extraoral tissues. The thyrocyte-expressed T2Rs control thyroid hormone production, and this regulatory role may be associated with susceptibility to thyroid diseases. This study examined whether the variations in TAS2Rs modify the risk of papillary thyroid carcinoma (PTC) and whether such T2R-related PTC risk is associated with genetically modifed thyroid function. We conducted a case-control study with 763 Korean females, including 250 PTC cases. Seventy-three single-nucleotide polymorphisms in 13 TAS2R genes and the pre-diagnosis levels of 4 thyroid-related functional markers [total triiodothyronine (TT3), free thyroxine, thyroid-stimulating hormone and thyroglobulin] were analysed. Individuals with TAS2R3/4 CC haplotype (rs2270009 and rs2234001) were at a lower risk for PTC than those with the remaining haplotypes (odds ratio = 0.59, 95% confdence interval: 0.36–0.97). Furthermore, TT3 levels were signifcantly reduced for TAS2R3/4 CC haplotype carriers compared with other haplotype carriers (p = 0.005). No other genetic variants exhibited critical associations with the PTC phenotype and biomarkers. In summary, genetic variations in T2R3/4 bitterness receptors may modify the PTC risk, and the genetically modifed thyroid hormone level by those variations may be linked with the PTC-T2Rs association. -

The Bitter Taste Receptor Tas2r14 Is Expressed in Ovarian Cancer and Mediates Apoptotic Signalling
THE BITTER TASTE RECEPTOR TAS2R14 IS EXPRESSED IN OVARIAN CANCER AND MEDIATES APOPTOTIC SIGNALLING by Louis T. P. Martin Submitted in partial fulfilment of the requirements for the degree of Master of Science at Dalhousie University Halifax, Nova Scotia June 2017 © Copyright by Louis T. P. Martin, 2017 DEDICATION PAGE To my grandparents, Christina, Frank, Brenda and Bernie, and my parents, Angela and Tom – for teaching me the value of hard work. ii TABLE OF CONTENTS LIST OF TABLES ............................................................................................................. vi LIST OF FIGURES .......................................................................................................... vii ABSTRACT ....................................................................................................................... ix LIST OF ABBREVIATIONS AND SYMBOLS USED .................................................... x ACKNOWLEDGEMENTS .............................................................................................. xii CHAPTER 1 INTRODUCTION ........................................................................................ 1 1.1 G-PROTEIN COUPLED RECEPTORS ................................................................ 1 1.2 GPCR CLASSES .................................................................................................... 4 1.3 GPCR SIGNALING THROUGH G PROTEINS ................................................... 6 1.4 BITTER TASTE RECEPTORS (TAS2RS) ........................................................... -

Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans?
nutrients Review Effects of Bitter Substances on GI Function, Energy Intake and Glycaemia-Do Preclinical Findings Translate to Outcomes in Humans? Peyman Rezaie †, Vida Bitarafan †, Michael Horowitz and Christine Feinle-Bisset * Adelaide Medical School and Centre of Research Excellence in Translating Nutritional Science to Good Health, Faculty of Health and Medical Sciences, University of Adelaide, Adelaide 5005, Australia; [email protected] (P.R.); [email protected] (V.B.); [email protected] (M.H.) * Correspondence: [email protected]; Tel.: +61-8-8313-6053 † These authors contributed equally to this work. Abstract: Bitter substances are contained in many plants, are often toxic and can be present in spoiled food. Thus, the capacity to detect bitter taste has classically been viewed to have evolved primarily to signal the presence of toxins and thereby avoid their consumption. The recognition, based on preclinical studies (i.e., studies in cell cultures or experimental animals), that bitter substances may have potent effects to stimulate the secretion of gastrointestinal (GI) hormones and modulate gut motility, via activation of bitter taste receptors located in the GI tract, reduce food intake and lower postprandial blood glucose, has sparked considerable interest in their potential use in the management or prevention of obesity and/or type 2 diabetes. However, it remains to be established whether findings from preclinical studies can be translated to health outcomes, including weight Citation: Rezaie, P.; Bitarafan, V.; loss and improved long-term glycaemic control. This review examines information relating to the Horowitz, M.; Feinle-Bisset, C. Effects effects of bitter substances on the secretion of key gut hormones, gastric motility, food intake and of Bitter Substances on GI Function, blood glucose in preclinical studies, as well as the evidence from clinical studies, as to whether Energy Intake and Glycaemia-Do findings from animal studies translate to humans. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Genome-Wide Association Study (GWAS) of Dental Caries in Diverse
Alotaibi et al. BMC Oral Health (2021) 21:377 https://doi.org/10.1186/s12903-021-01670-5 RESEARCH Open Access Genome-Wide Association Study (GWAS) of dental caries in diverse populations Rasha N. Alotaibi1,2,3*, Brian J. Howe4,5, Jonathan M. Chernus1,6, Nandita Mukhopadhyay1,2, Carla Sanchez1,2, Frederic W. B. Deleyiannis7, Katherine Neiswanger1,2, Carmencita Padilla8, Fernando A. Poletta9, Ieda M. Orioli10, Carmen J. Buxó11, Jacqueline T. Hecht12, George L. Wehby13, Ross E. Long14, Alexandre R. Vieira1, Seth M. Weinberg1,2, John R. Shafer1,2,6, Lina M. Moreno Uribe4,15 and Mary L. Marazita1,2,6 Abstract Background: Dental caries is one of the most common chronic diseases and is infuenced by a complex interplay of genetic and environmental factors. Most previous genetic studies of caries have focused on identifying genes that contribute to dental caries in specifc ethnic groups, usually of European descent. Methods: The aim of this study is to conduct a genome-wide association study (GWAS) to identify associations afecting susceptibility to caries in a large multiethnic population from Argentina, the Philippines, Guatemala, Hun- gary, and the USA, originally recruited for studies of orofacial clefts (POFC, N 3686). Ages of the participants ranged from 2 to 12 years for analysis of the primary dentition, and 18–60 years for analysis= of the permanent dentition. For each participant, dental caries was assessed by counts of decayed and flled teeth (dft/DFT) and genetic variants (single nucleotide polymorphisms, SNPs) were genotyped or imputed across the entire genome. Caries was analyzed separately for the primary and permanent dentitions, with age, gender, and presence/absence of any type of OFC treated as covariates. -
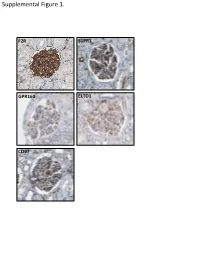
Supplemental Data
Supplemental Figure 1. F2R S1PR1 GPR160 ELTD1 CD97 Supplemental Figure 2. A B brain heart lung liver kidney spleen testis skm Expression in mouse ssues glom rok Gprc5a Gapdh Human Protein Atlas RNAseq database Supplemental Figure 3. A Gprc5a Pdgfrb Merged CL CL Gprc5a CD31 Merged CL CL B C 1.4 * 1.2 mc 1 matrix 0.8 0.6 0.4 matrix 0.2 0 pod end 1 2 mes3 D Vector Gprc5a E 40 kD - - Gprc5a - acn Supplemental Figure 4. Control 12 month-old KO 12 month-old A B C D E F 1 2 0.8 * 1.5 score 0.6 m µ 1 0.4 0.2 Slits/ 0.5 Mesangial 0 0 Ctrl KO Ctrl KO Supplemental Figure 5. A B 1.2 Vector Gprc5a 1 * 0.8 0.6 * 0.4 * Normalized Density 0.2 0 pEGFR/tEGR pSmad/tSmad TGF-β1 C 1.8 siCON 1.6 siGprc5a * 1.4 * * 1.2 1 0.8 0.6 NormalizedDensity 0.4 0.2 0 pEGFR/tEGR pSmad/tSmad TGF-β1 Supplemental table 1. List of glomerulus-expressed GPCRs as detected by qPCR. Data shown as mean ± standard deviation (Glom=glomerulus, Rok=rest of kidney). GPCR Glom Rok Glom/Rok LPAR6 41892,11 ± 38478,89 1040,06 ± 1370,12 39,28 ELTD1 30275,64 ± 14085,26 33,23 ± 46,99 910,13 GPR116 24020,06 ± 7789,84 55,1 ± 47,75 434,96 PTH1R 15402,81 ± 17644,32 7521,01 ± 3264,57 1,05 CALCRL 14096,09 ± 3854,84 199,06 ± 222,52 69,81 HPRT1 13342,04 ± 10677,69 1824,77 ± 1767,23 6,31 S1PR5 9474,7 ± 10124,3 110,84 ± 29,54 84,48 LPHN2 8645,89 ± 914,74 256,04 ± 293,87 32,77 FZD1 8176,2 ± 4947,45 1321,97 ± 1311,91 5,18 CXCR4 7097,31 ± 4388,91 535,98 ± 640,28 12,24 GPR160 6446,59 ± 1550,07 4816,3 ± 5918,99 0,34 NPY1R 6177,74 ± 6282,76 209,64 ± 296,48 28,47 PTGER4 5323,66 ± 3789,51 179,13 ± 210 28,72 RXFP1 -

Variation in Ligand Responses of the Bitter Taste Receptors TAS2R1 And
Tsutsui et al. BMC Evolutionary Biology (2016) 16:208 DOI 10.1186/s12862-016-0783-0 RESEARCH ARTICLE Open Access Variation in ligand responses of the bitter taste receptors TAS2R1 and TAS2R4 among New World monkeys Kei Tsutsui1, Masahiro Otoh2, Kodama Sakurai2, Nami Suzuki-Hashido1, Takashi Hayakawa1,3, Takumi Misaka4, Yoshiro Ishimaru4, Filippo Aureli5,6, Amanda D. Melin7, Shoji Kawamura2* and Hiroo Imai1* Abstract Background: New World monkeys (NWMs) are unique in that they exhibit remarkable interspecific variation in color vision and feeding behavior, making them an excellent model for studying sensory ecology. However, it is largely unknown whether non-visual senses co-vary with feeding ecology, especially gustation, which is expected to be indispensable in food selection. Bitter taste, which is mediated by bitter taste receptors (TAS2Rs) in the tongue, helps organisms avoid ingesting potentially toxic substances in food. In this study, we compared the ligand sensitivities of the TAS2Rs of five species of NWMs by heterologous expression in HEK293T cells and calcium imaging. Results: We found that TAS2R1 and TAS2R4 orthologs differ in sensitivity among the NWM species for colchicine and camphor, respectively. We then reconstructed the ancestral receptors of NWM TAS2R1 and TAS2R4, measured the evolutionary shift in ligand sensitivity, and identified the amino acid replacement at residue 62 as responsible for the high sensitivity of marmoset TAS2R4 to colchicine. Conclusions: Our results provide a basis for understanding the differences in feeding ecology among NWMs with respect to bitter taste. Keywords: Bitter taste receptor, TAS2R, G protein-coupled receptor, New World monkey, Interspecific functional variation, Molecular evolution Background of TAS2Rs among species.