A Machine Learning Workflow Performing Virtual Knockout Experiments on Single-Cell Gene Regulatory Networks
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening and Identification of Key Biomarkers in Clear Cell Renal Cell Carcinoma Based on Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Screening and identification of key biomarkers in clear cell renal cell carcinoma based on bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 , Iranna Kotturshetti 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. 3. Department of Ayurveda, Rajiv Gandhi Education Society`s Ayurvedic Medical College, Ron, Karnataka 562209, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.21.423889; this version posted December 23, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Clear cell renal cell carcinoma (ccRCC) is one of the most common types of malignancy of the urinary system. The pathogenesis and effective diagnosis of ccRCC have become popular topics for research in the previous decade. In the current study, an integrated bioinformatics analysis was performed to identify core genes associated in ccRCC. An expression dataset (GSE105261) was downloaded from the Gene Expression Omnibus database, and included 26 ccRCC and 9 normal kideny samples. Assessment of the microarray dataset led to the recognition of differentially expressed genes (DEGs), which was subsequently used for pathway and gene ontology (GO) enrichment analysis. -

Atlas Antibodies in Breast Cancer Research Table of Contents
ATLAS ANTIBODIES IN BREAST CANCER RESEARCH TABLE OF CONTENTS The Human Protein Atlas, Triple A Polyclonals and PrecisA Monoclonals (4-5) Clinical markers (6) Antibodies used in breast cancer research (7-13) Antibodies against MammaPrint and other gene expression test proteins (14-16) Antibodies identified in the Human Protein Atlas (17-14) Finding cancer biomarkers, as exemplified by RBM3, granulin and anillin (19-22) Co-Development program (23) Contact (24) Page 2 (24) Page 3 (24) The Human Protein Atlas: a map of the Human Proteome The Human Protein Atlas (HPA) is a The Human Protein Atlas consortium cell types. All the IHC images for Swedish-based program initiated in is mainly funded by the Knut and Alice the normal tissue have undergone 2003 with the aim to map all the human Wallenberg Foundation. pathology-based annotation of proteins in cells, tissues and organs expression levels. using integration of various omics The Human Protein Atlas consists of technologies, including antibody- six separate parts, each focusing on References based imaging, mass spectrometry- a particular aspect of the genome- 1. Sjöstedt E, et al. (2020) An atlas of the based proteomics, transcriptomics wide analysis of the human proteins: protein-coding genes in the human, pig, and and systems biology. mouse brain. Science 367(6482) 2. Thul PJ, et al. (2017) A subcellular map of • The Tissue Atlas shows the the human proteome. Science. 356(6340): All the data in the knowledge resource distribution of proteins across all eaal3321 is open access to allow scientists both major tissues and organs in the 3. -

View Is Portrayed Schematically in Figure 7B
BASIC RESEARCH www.jasn.org Recombination Signal Binding Protein for Ig-kJ Region Regulates Juxtaglomerular Cell Phenotype by Activating the Myo-Endocrine Program and Suppressing Ectopic Gene Expression † † ‡ Ruth M. Castellanos-Rivera,* Ellen S. Pentz,* Eugene Lin,* Kenneth W. Gross, † Silvia Medrano,* Jing Yu,§ Maria Luisa S. Sequeira-Lopez,* and R. Ariel Gomez* *Department of Pediatrics, School of Medicine, †Department of Biology, Graduate School of Arts and Sciences, and §Department of Cell Biology, University of Virginia, Charlottesville, Virginia; and ‡Department of Molecular and Cellular Biology, Roswell Park Cancer Institute, Buffalo, New York ABSTRACT Recombination signal binding protein for Ig-kJ region (RBP-J), the major downstream effector of Notch signaling, is necessary to maintain the number of renin-positive juxtaglomerular cells and the plasticity of arteriolar smooth muscle cells to re-express renin when homeostasis is threatened. We hypothesized that RBP-J controls a repertoire of genes that defines the phenotype of the renin cell. Mice bearing a bacterial artificial chromosome reporter with a mutated RBP-J binding site in the renin promoter had markedly reduced reporter expression at the basal state and in response to a homeostatic challenge. Mice with conditional deletion of RBP-J in renin cells had decreased expression of endocrine (renin and Akr1b7)and smooth muscle (Acta2, Myh11, Cnn1,andSmtn) genes and regulators of smooth muscle expression (miR- 145, SRF, Nfatc4, and Crip1). To determine whether RBP-J deletion decreased the endowment of renin cells, we traced the fate of these cells in RBP-J conditional deletion mice. Notably, the lineage staining patterns in mutant and control kidneys were identical, although mutant kidneys had fewer or no renin- expressing cells in the juxtaglomerular apparatus. -

Page 1 Supplemental Table I Upin WTVG Upin Humlowfb FAR1
Supplemental Table I UPinWTvG UPinHuMlowFB overlap FAR1 CD209 PLEKHO2 B2M IL27 GCLC CALR HMGN2P46 ME1 XPO1 CLEC1A NEK6 PDIA3 HSD17B14 TMEM106A SERPINH1 NSUN7 KCNJ15 PLEKHO2 SEMA6B SH3PXD2B HSPD1 BAALC RHOU SYVN1 CEACAM4 TGFBI DNAJB9 KATNAL2 CTSZ SLC25A19 CCDC175 SULF2 MHCII CARD14 P2RY6 MDN1 TDO2 CD74 GCLC FCAR HLA-DQB1 PTPN2 GLDN CD14 CHORDC1 MMP7 CSF2RB LOX CLEC5A MRC1 STIP1 ZMYND15 ITGAX BPIFB1 ITLN1 SLAMF7 ME1 DKK2 CD84 PDIA6 FAM124A FCGR2A HYOU1 F3 CLEC10A NLRC5 DZIP1L IFI30 NEK6 CECR6 CLEC4A SLC39A14 NDP CLEC7A TMEM106AFCGR1A TFEC KCNJ15 CCL7 C1QC SH3PXD2B CRABP2 C1QA RHOU PIPOX FOLR2 LRP2 CCL2 CH25H MVD VSIG4 C1QB TGFBI LINC01010 SIGLEC1 NFKB2 DPRXP4 CTSS C5 FAM20A CCR1 HSP90B1 ANKRD29 SLAMF8 ALDH18A1 OCSTAMP MS4A7 EDEM1 TGM2 HK3 CTSZ TM4SF19 CXCL10 C6 TRPV4 CTSK AACS CCL8 MSR1 PPA1 CCL1 STEAP4 PIK3R5 KCNE1 CXCL9 RASAL3 SLC9A7P1 MS4A6A DOCK11 CHI3L1 TIMP1 BHLHE40 LOC731424 CD209 HCLS1 DCSTAMP CCL7 FASN MSR1 PDCD1LG2 PDIA4 IL31RA CCL2 ITK CXCL3 CXCL2 CRELD2 TREM2 C15orf48 SFTPD MGST1 GPR84 BCL3 METTL7B CXCL5 SULF2 TMEM86A OCSTAMP CYP51A1 A2M SERPINA1 CREB3L1 AQP9 MMP12 DUSP2 NUPR1 CCL8 ADAM8 FHAD1 CCL24 P2RY6 YPEL4 FBP1 KCNAB2 FBP1 NA NFKBIE LOC100506585 NA FSCN1 CXCL16 NA MANF RAB13 NA SLC5A3 LOC391322 NA CTSC IL8 NA COTL1 MS4A4A NA HSPA5 SERPING1 NA MUC5B PLA2G4C NA CD74 CA12 NA HLA-DQB1 GBP1P1 NA SLC7A2 C11orf45 NA FABP5 ACVRL1 NA CIITA SPP1 NA RAB3IL1 TLN2 NA HSPE1 NDRG2 NA SCD C15orf48 NA ITIH4 KCNJ15 NA SERPINA3 MEIS3P1 NA LAG3 IL1RN NA FOXM1 HNMT NA CD14 CYP27B1 NA RRM2 CDCP1 NA ABCD2 FOLR2 NA FCRL2 ECM1 NA PDE3B ADAMDEC1 -

(12) Patent Application Publication (10) Pub. No.: US 2014/0304845 A1 Loboda Et Al
US 201403.04845A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0304845 A1 Loboda et al. (43) Pub. Date: Oct. 9, 2014 54) ALZHEMIERS DISEASE SIGNATURE Publicationublication ClassificatiClassification MARKERS AND METHODS OF USE (51) Int. Cl. (71) Applicant: MERCKSHARP & DOHME CORP, CI2O I/68 (2006.01) Rahway, NJ (US) AOIK 67/027 (2006.01) (52) U.S. Cl. (72) Inventors: SIES yet.sS); CPC .......... CI2O I/6883 (2013.01); A0IK 67/0275 Icnaei NepoZnyn, Yeadon, (2013.01) le.East,Italia; David J. Stone, Wyncote, USPC .............. 800/12:536/23.5:536/23.2:506/17 (US); Keith Tanis, Quakertown, PA (US); William J. Ray, Juniper, FL (US) (57) ABSTRACT (21) Appl. No.: 14/354,622 Methods, biomarkers, and expression signatures are dis 1-1. closed for assessing the disease progression of Alzheimer's (22) PCT Filed: Oct. 26, 2012 disease (AD). In one embodiment, BioAge (biological age), NdStress (neurodegenerative stress), Alz (Alzheimer), and (86). PCT No.: PCT/US12A62218 Inflame (inflammation) are used as biomarkers of AD pro S371 (c)(1), gression. In another aspect, the invention comprises a gene (2), (4) Date: Apr. 28, 2014 signature for evaluating disease progression. In still another Related U.S. Application Data abiliticises in (60) Provisional application No. 61/553,400, filed on Oct. used to identify animal models for use in the development and 31, 2011. evaluation of therapeutics for the treatment of AD. Patent Application Publication Oct. 9, 2014 Sheet 1 of 16 US 2014/0304845 A1 Y : : O O O O O O O O D O v v CN On cy Patent Application Publication Oct. -

MS4A7 (NM 206939) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP312066 MS4A7 (NM_206939) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human membrane-spanning 4-domains, subfamily A, member 7 (MS4A7), transcript variant 3 Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 26 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_996822 Locus ID: 58475 UniProt ID: Q9GZW8, A0A024R556 RefSeq Size: 2976 Cytogenetics: 11q12.2 RefSeq ORF: 720 Synonyms: 4SPAN2; CD20L4; CFFM4; MS4A8 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 MS4A7 (NM_206939) Human Recombinant Protein – TP312066 Summary: This gene encodes a member of the membrane-spanning 4A gene family, members of which are characterized by common structural features and similar intron/exon splice boundaries and display unique expression patterns in hematopoietic cells and nonlymphoid tissues. This family member is associated with mature cellular function in the monocytic lineage, and it may be a component of a receptor complex involved in signal transduction. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -
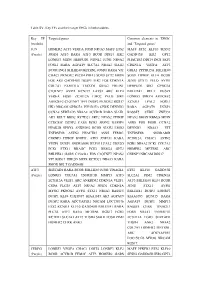
Targeted Genes Common Elements In
Table SV. Key TFs and their target DEGs in hub modules. Key TF Targeted genes Common elements in ‘DEGs’ (module) and ‘Targeted genes’ JUN HOMER2 ATF3 VEGFA FOSB NR4A3 MAFF ETS2 MAFF ETS2 KLF10 SESN2 (Purple) JOSD1 ATF3 RARA ATF3 BCOR DDIT4 IER2 GADD45B IER2 GPT2 LONRF3 MIDN HERPUD1 NDNL2 JUNB NR4A2 PHACTR3 DDIT4 ING1 SKP2 FOSL1 RARA AGPAT9 SLC7A1 NR4A2 SIAH2 CDKN1A VEGFA ATF3 BCOR ING1 BHLHE40 METRNL JOSD1 RARA VIT GRIA2 PPP1R15A BHLHE40 CHAC1 PKNOX2 PLCB4 PHF13 SOX9 SYT2 MIDN SOX9 ITPRIP KLF4 BCOR FOS AK5 GADD45B DUSP1 IER2 FOS CDKN1A JUNB STX11 PELO AVPI1 COL7A1 FAM131A TBCCD1 GRIA2 ERLIN1 HERPUD1 SIK1 GPRC5A C1QTNF7 AVPI1 KCNJ15 LATS2 ARC KLF4 BHLHE41 RELT DUSP2 VEGFA FOSB ZC3H12A LMO2 PELO SIK1 LONRF3 SPRY4 ARHGEF2 ARHGEF2 C1QTNF7 TFPI DUSP2 PKNOX2 RGS17 KCNJ15 LPAL2 FOSL1 HK1 NRCAM GPRC5A PPP1R15A CERK DENND3 RARA AGPAT9 DUSP1 CCNA2 SERTAD3 NR4A2 ACVR1B RARA SIAH1 RASSF5 CERK ZNF331 AK5 RELT BRD2 KCTD21 SKP2 NPAS2 ITPRIP NPAS2 SMOX RBM24 MIDN CCDC85C DUSP2 CARS EGR1 SESN2 RASSF5 ASNS FOS FOSB CCNA2 PIP4K2B SPRY4 ANKRD52 BCOR SIAH2 LMO2 DENND3 NR4A3 VIT TNFRSF1B ASTN2 PHACTR3 ASNS FEM1C TNFRSF1B SNORA80B CSRNP3 ITPRIP BNIP3L ATF3 ZNF331 RARA ZC3H12A CHAC1 ASTN2 VEZF1 DUSP1 SNORA80B KLF10 LPAL2 UBE2O EGR1 NR4A2 PCK1 COL7A1 PCK1 STX11 RRAGC PCK1 RBM24 GPT2 HOMER2 METRNL ARC BHLHE41 GARS C15orf41 FOS C1QTNF7 NPAS2 CSRNP3 NRCAM RGS17 VIT RGS17 UBE2O SOX9 KCTD21 NR4A3 RARA SMOX RELT GADD45B ATF3 SERTAD1 RARA BCOR BHLHE40 JUNB TINAGL1 ETS2 KLF10 GADD45B (Purple) LONRF3 COL7A1 TNFRSF1B MMP13 ATF3 SLC2A1 PIM2 CDKN1A ZC3H12A VEZF1 ARC ANKRD52 -

Mapping Human Hematopoietic Hierarchy at Single Cell Resolution by Microwell-Seq
bioRxiv preprint doi: https://doi.org/10.1101/127217; this version posted April 13, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Mapping Human Hematopoietic Hierarchy at Single Cell Resolution by Microwell-seq Shujing Lai1,8,9, Yang Xu1,8,9, Wentao Huang1,9, Mengmeng Jiang1,8, 9, Haide Chen1,8, Fang Ye1, Renying Wang1, Yunfei Qiu 2, Xinyi Jiang1, Daosheng Huang1,8, Jie Mao3, Yanwei Li 4, Yingru Lu5, Jin Xie6, Qun Fang7,Tiefeng Li3, He Huang2,8, Xiaoping Han1,8 & Guoji Guo1,8* 1Center for Stem Cell and Regenerative Medicine, Zhejiang University School of Medicine, Hangzhou 310058, China 2The 1st Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China 3Institute of Applied Mechanics, Zhejiang University, Hangzhou 310027, China 4Core Facilities, Zhejiang University School of Medicine, Hangzhou 310058, China 5Department of Emergency Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325000, China 6Institute of Mechatronic Control Engineering, Zhejiang University, Hangzhou 310027, China 7Institute of Microanalytical Systems, Department of Chemistry, Zhejiang University, Hangzhou, 310058, China 8Stem Cell Institute, Zhejiang University, Hangzhou 310058, China 9These authors contributed equally to this work * Correspondence: (G.G.) [email protected]; Summary: The classical hematopoietic hierarchy, which is mainly built with fluorescence-activated cell sorting (FACS) technology, proves to be inaccurate in recent studies. Single cell RNA-seq (scRNA-seq) analysis provides a solution to overcome the limit of FACS-based cell type definition system for the dissection of complex cellular hierarchy. -

Regulation of Cancer Stemness in Breast Ductal Carcinoma in Situ by Vitamin D Compounds
Author Manuscript Published OnlineFirst on May 28, 2020; DOI: 10.1158/1940-6207.CAPR-19-0566 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Analysis of the Transcriptome: Regulation of Cancer Stemness in Breast Ductal Carcinoma In Situ by Vitamin D Compounds Naing Lin Shan1, Audrey Minden1,5, Philip Furmanski1,5, Min Ji Bak1, Li Cai2,5, Roman Wernyj1, Davit Sargsyan3, David Cheng3, Renyi Wu3, Hsiao-Chen D. Kuo3, Shanyi N. Li3, Mingzhu Fang4, Hubert Maehr1, Ah-Ng Kong3,5, Nanjoo Suh1,5 1Department of Chemical Biology, Ernest Mario School of Pharmacy; 2Department of Biomedical Engineering, School of Engineering; 3Department of Pharmaceutics, Ernest Mario School of Pharmacy; 4Environmental and Occupational Health Sciences Institute and School of Public Health, 5Rutgers Cancer Institute of New Jersey, New Brunswick; Rutgers, The State University of New Jersey, NJ, USA Running title: Regulation of cancer stemness by vitamin D compounds Key words: Breast cancer, cancer stemness, gene expression, DCIS, vitamin D compounds Financial Support: This research was supported by the National Institutes of Health grant R01 AT007036, R01 AT009152, ES005022, Charles and Johanna Busch Memorial Fund at Rutgers University and the New Jersey Health Foundation. Corresponding author: Dr. Nanjoo Suh, Department of Chemical Biology, Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, 164 Frelinghuysen Road, Piscataway, New Jersey 08854. Tel: 848-445-8030, Fax: 732-445-0687; e-mail: [email protected] Disclosure of Conflict of Interest: “The authors declare no potential conflicts of interest” 1 Downloaded from cancerpreventionresearch.aacrjournals.org on October 1, 2021. -

Single-Cell RNA Sequencing Identifies Candidate Renal
BASIC RESEARCH www.jasn.org Single-Cell RNA Sequencing Identifies Candidate Renal Resident Macrophage Gene Expression Signatures across Species Kurt A. Zimmerman,1 Melissa R. Bentley,1 Jeremie M. Lever ,2 Zhang Li,1 David K. Crossman,3 Cheng Jack Song,1 Shanrun Liu,4 Michael R. Crowley,3 James F. George,5 Michal Mrug,2,6 and Bradley K. Yoder1 1Department of Cell, Developmental, and Integrative Biology, 2Division of Nephrology, Department of Medicine, 3Department of Genetics, 4Department of Biochemistry and Molecular Genetics, and 5Division of Cardiothoracic Surgery, Department of Surgery, University of Alabama at Birmingham, Birmingham, Alabama; and 6Department of Veterans Affairs Medical Center, Birmingham, Alabama ABSTRACT Background Resident macrophages regulate homeostatic and disease processes in multiple tissues, in- cluding the kidney. Despite having well defined markers to identify these cells in mice, technical limita- tions have prevented identification of a similar cell type across species. The inability to identify resident macrophage populations across species hinders the translation of data obtained from animal model to human patients. Methods As an entry point to determine novel markers that could identify resident macrophages across species, we performed single-cell RNA sequencing (scRNAseq) analysis of all T and B cell–negative CD45+ innate immune cells in mouse, rat, pig, and human kidney tissue. Results We identified genes with enriched expression in mouse renal resident macrophages that were also present in candidate resident macrophage populations across species. Using the scRNAseq data, we defined a novel set of possible cell surface markers (Cd74 and Cd81) for these candidate kidney resident macrophages. We confirmed, using parabiosis and flow cytometry, that these proteins are indeed enriched in mouse resident macrophages. -

Flavone Effects on the Proteome and Transcriptome of Colonocytes in Vitro and in Vivo and Its Relevance for Cancer Prevention and Therapy
TECHNISCHE UNIVERSITÄT MÜNCHEN Lehrstuhl für Ernährungsphysiologie Flavone effects on the proteome and transcriptome of colonocytes in vitro and in vivo and its relevance for cancer prevention and therapy Isabel Winkelmann Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. D. Haller Prüfer der Dissertation: 1. Univ.-Prof. Dr. H. Daniel 2. Univ.-Prof. Dr. U. Wenzel (Justus-Liebig-Universität Giessen) 3. Prof. Dr. E.C.M. Mariman (Maastricht University, Niederlande) schriftliche Beurteilung Die Dissertation wurde am 24.08.2009 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 25.11.2009 angenommen. Die Forschung ist immer auf dem Wege, nie am Ziel. (Adolf Pichler) Table of contents 1. Introduction .......................................................................................................... 1 1.1. Cancer and carcinogenesis .................................................................................. 2 1.2. Colorectal Cancer ............................................................................................... 3 1.2.1. Hereditary forms of CRC ........................................................................................ 4 1.2.2. Sporadic forms of CRC ..........................................................................................