Mrna Expression Profiles Obtained from Microdissected Pancreatic Cancer Cells Can Predict Patient Survival
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical, Molecular, and Immune Analysis of Dabrafenib-Trametinib
Supplementary Online Content Chen G, McQuade JL, Panka DJ, et al. Clinical, molecular and immune analysis of dabrafenib-trametinib combination treatment for metastatic melanoma that progressed during BRAF inhibitor monotherapy: a phase 2 clinical trial. JAMA Oncology. Published online April 28, 2016. doi:10.1001/jamaoncol.2016.0509. eMethods. eReferences. eTable 1. Clinical efficacy eTable 2. Adverse events eTable 3. Correlation of baseline patient characteristics with treatment outcomes eTable 4. Patient responses and baseline IHC results eFigure 1. Kaplan-Meier analysis of overall survival eFigure 2. Correlation between IHC and RNAseq results eFigure 3. pPRAS40 expression and PFS eFigure 4. Baseline and treatment-induced changes in immune infiltrates eFigure 5. PD-L1 expression eTable 5. Nonsynonymous mutations detected by WES in baseline tumors This supplementary material has been provided by the authors to give readers additional information about their work. © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/30/2021 eMethods Whole exome sequencing Whole exome capture libraries for both tumor and normal samples were constructed using 100ng genomic DNA input and following the protocol as described by Fisher et al.,3 with the following adapter modification: Illumina paired end adapters were replaced with palindromic forked adapters with unique 8 base index sequences embedded within the adapter. In-solution hybrid selection was performed using the Illumina Rapid Capture Exome enrichment kit with 38Mb target territory (29Mb baited). The targeted region includes 98.3% of the intervals in the Refseq exome database. Dual-indexed libraries were pooled into groups of up to 96 samples prior to hybridization. -

An Analysis About Heterogeneity Among Cancers Based on the DNA Methylation Patterns
An analysis about heterogeneity among cancers based on the DNA Methylation Patterns Yang Liu Harbin Institute of Technology Yue Gu Harbin Medical University Mu Su Harbin Institute of Technology Hui Liu Harbin Institute of Technology Shumei Zhang Harbin Medical University Yan Zhang ( [email protected] ) Harbin Institute of Technology https://orcid.org/0000-0002-5307-2484 Research article Keywords: DNA methylation, cancer, epigenetic heterogeneity, survival analysis Posted Date: July 17th, 2019 DOI: https://doi.org/10.21203/rs.2.11636/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at BMC Cancer on December 1st, 2019. See the published version at https://doi.org/10.1186/s12885-019-6455-x. Page 1/28 Abstract Background: The occurrence of cancer is usually the result of a co-effect of genetic and environmental factors. It is generally believed that the main cause of cancer is the accumulation of genetic mutations, and DNA methylation, as one of the epigenetic modications closely related to environmental factors, participates in the regulation of gene expression and cell differentiation and plays an important role in the development of cancer. Methods: This article discusses the epigenetic heterogeneity of cancer in detail. Firstly DNA methylation data of 7 cancer types were obtained from Illumina Innium HumanMethylation 450K platform of TCGA database. Diagnostic markers of each cancer were obtained by t-test and absolute difference of DNA differencial methylation analysis. Enrichment analysis of these specic markers indicated that they were involved in different biological functions. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

OR6M1 (NM 001005325) Human Tagged ORF Clone – RG214544
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RG214544 OR6M1 (NM_001005325) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: OR6M1 (NM_001005325) Human Tagged ORF Clone Tag: TurboGFP Symbol: OR6M1 Synonyms: OR11-271 Vector: pCMV6-AC-GFP (PS100010) E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RG214544 representing NM_001005325 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGGAAACTGGAGCACTGTGACTGAAATCACCCTAATTGCCTTCCCAGCTCTCCTGGAGATTCGAATAT CTCTCTTCGTGGTTCTTGTGGTAACTTACACATTAACAGCAACAGGAAACATCACCATCATCTCCCTGAT ATGGATTGATCATCGCCTGCAAACTCCAATGTACTTCTTCCTCAGTAATTTGTCCTTTCTGGATATCTTA TACACCACTGTCATTACCCCAAAGTTGTTGGCCTGCCTCCTAGGAGAAGAGAAAACCATATCTTTTGCTG GTTGCATGATCCAAACATATTTCTACTTCTTTCTGGGGACGGTGGAGTTTATCCTCTTGGCGGTGATGTC CTTTGACCGCTACATGGCTATCTGCGACCCACTGCACTACACGGTCATCATGAACAGCAGGGCCTGCCTT CTGCTGGTTCTGGGATGCTGGGTGGGAGCCTTCCTGTCTGTGTTGTTTCCAACCATTGTAGTGACAAGGC TACCTTACTGTAGGAAAGAAATTAATCATTTCTTCTGTGACATTGCCCCTCTTCTTCAGGTGGCCTGTAT AAATACTCACCTCATTGAGAAGATAAACTTTCTCCTCTCTGCCCTTGTCATCCTGAGCTCCCTGGCATTC ACTACTGGGTCCTACGTGTACATAATTTCTACCATCCTGCGTATCCCCTCCACCCAGGGCCGTCAGAAAG CTTTTTCTACCTGTGCTTCTCACATCACTGTTGTCTCCATTGCCCACGGGAGCAACATCTTTGTGTATGT GAGACCCAATCAGAACTCCTCACTGGATTATGACAAGGTGGCCGCTGTCCTCATCACAGTGGTGACCCCT CTCCTGAACCCTTTTATCTACAGCTTGAGGAATGAGAAGGTACAGGAAGTGTTGAGAGAGACAGTGAACA -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

INTRODUCING a NOVEL METHOD for GENETIC ANALYSIS of AUTISM SPECTRUM DISORDER Sepideh Nouri
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 12-2013 INTRODUCING A NOVEL METHOD FOR GENETIC ANALYSIS OF AUTISM SPECTRUM DISORDER sepideh nouri Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Computational Biology Commons, and the Genomics Commons Recommended Citation nouri, sepideh, "INTRODUCING A NOVEL METHOD FOR GENETIC ANALYSIS OF AUTISM SPECTRUM DISORDER" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 417. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/417 This Thesis (MS) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. INTRODUCING A NOVEL METHOD FOR GENETIC ANALYSIS OF AUTISM SPECTRUM DISORDER by Sepideh Nouri, M.S. APPROVED: Eric Boerwinkle, Ph.D., Supervisor Kim-Anh Do, Ph.D. Alanna Morrison, Ph.D. James Hixson, Ph.D. Paul Scheet, Ph.D. APPROVED: Dean, The University of Texas Graduate School of Biomedical Sciences at Houston INTRODUCING A NOVEL METHOD FOR GENETIC ANALYSIS OF AUTISM SPECTRUM DISORDER A THESIS Presented to the Faculty of The University of Texas Health Science Center at Houston and The University of Texas M. -
An Analysis About Heterogeneity Among Cancers Based on the DNA Methylation Patterns Yang Liu1, Yue Gu2,Musu1, Hui Liu2, Shumei Zhang3* and Yan Zhang1*
Liu et al. BMC Cancer (2019) 19:1259 https://doi.org/10.1186/s12885-019-6455-x RESEARCH ARTICLE Open Access An analysis about heterogeneity among cancers based on the DNA methylation patterns Yang Liu1, Yue Gu2,MuSu1, Hui Liu2, Shumei Zhang3* and Yan Zhang1* Abstract Background: It is generally believed that DNA methylation, as one of the most important epigenetic modifications, participates in the regulation of gene expression and plays an important role in the development of cancer, and there exits epigenetic heterogeneity among cancers. Therefore, this study tried to screen for reliable prognostic markers for different cancers, providing further explanation for the heterogeneity of cancers, and more targets for clinical transformation studies of cancer from epigenetic perspective. Methods: This article discusses the epigenetic heterogeneity of cancer in detail. Firstly, DNA methylation data of seven cancer types were obtained from Illumina Infinium HumanMethylation 450 K platform of TCGA database. Then, differential methylation analysis was performed in the promotor region. Secondly, pivotal gene markers were obtained by constructing the DNA methylation correlation network and the gene interaction network in the KEGG pathway, and 317 marker genes obtained from two networks were integrated as candidate markers for the prognosis model. Finally, we used the univariate and multivariate COX regression models to select specific independent prognostic markers for each cancer, and studied the risk factor of these genes by doing survival analysis. Results: First, the cancer type-specific gene markers were obtained by differential methylation analysis and they were found to be involved in different biological functions by enrichment analysis. Moreover, specific and common diagnostic markers for each type of cancer was sorted out and Kaplan-Meier survival analysis showed that there was significant difference in survival between the two risk groups. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -
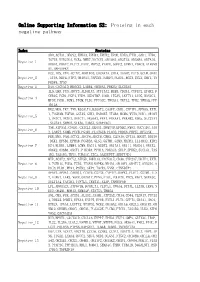
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -
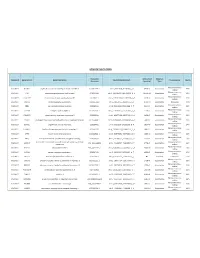
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein) -

A Mini-Review
omics & en P g h o a c r a m m a Abaffy, J Pharmacogenomics Pharmacoproteomics 2015, 6:4 r c a o Journal of h p P r o f DOI: 10.4172/2153-0645.1000152 t o e l ISSN: 2153-0645o a m n r i c u s o J Pharmacogenomics & Pharmacoproteomics Review Open Access Human Olfactory Receptors Expression and Their Role in Non-Olfactory Tissues – A Mini-Review Tatjana Abaffy* Department of Molecular and Cellular Pharmacology, University of Miami, Miami, USA *Corresponding author: Tatjana Abaffy, Department of Molecular and Cellular Pharmacology, University of Miami, Miami, USA, Tel: 305 243 1508; E-mail: [email protected] Received date: September 07, 2015; Accepted date: October 01, 2015; Published date: October 06, 2015 Copyright: © 2015 Abaffy T. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract The expression of human olfactory receptors in non-olfactory tissues has been documented since the early nineties, however, until recently their functional roles were largely unknown. Many studies have demonstrated that these G-protein coupled receptors (GPCRs) are actively involved in various cellular processes. Here, we summarized current evidence describing the most prominent expression and functional data for these ectopic olfactory receptors. Further studies focused on discovering their ligands, both agonists and antagonists, will be necessary to fully characterize molecular mechanisms underlying their functional roles in human physiology and pathophysiology. Keywords: Olfactory receptors; GPCRs; Ectopic expression The existence of olfactory receptors outside the olfactory sensory system was first documented in mammalian germ cells, and it was Introduction suggested that these “ectopic” ORs could have a role in chemotaxis during fertilization [7-9]. -

Supplementary Methods
doi: 10.1038/nature06162 SUPPLEMENTARY INFORMATION Supplementary Methods Cloning of human odorant receptors 423 human odorant receptors were cloned with sequence information from The Olfactory Receptor Database (http://senselab.med.yale.edu/senselab/ORDB/default.asp). Of these, 335 were predicted to encode functional receptors, 45 were predicted to encode pseudogenes, 29 were putative variant pairs of the same genes, and 14 were duplicates. We adopted the nomenclature proposed by Doron Lancet 1. OR7D4 and the six intact odorant receptor genes in the OR7D4 gene cluster (OR1M1, OR7G2, OR7G1, OR7G3, OR7D2, and OR7E24) were used for functional analyses. SNPs in these odorant receptors were identified from the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) or through genotyping. OR7D4 single nucleotide variants were generated by cloning the reference sequence from a subject or by inducing polymorphic SNPs by site-directed mutagenesis using overlap extension PCR. Single nucleotide and frameshift variants for the six intact odorant receptors in the same gene cluster as OR7D4 were generated by cloning the respective genes from the genomic DNA of each subject. The chimpanzee OR7D4 orthologue was amplified from chimpanzee genomic DNA (Coriell Cell Repositories). Odorant receptors that contain the first 20 amino acids of human rhodopsin tag 2 in pCI (Promega) were expressed in the Hana3A cell line along with a short form of mRTP1 called RTP1S, (M37 to the C-terminal end), which enhances functional expression of the odorant receptors 3. For experiments with untagged odorant receptors, OR7D4 RT and S84N variants without the Rho tag were cloned into pCI.