Table of Contents List of Investigators
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands
The Activation of the Glucagon-Like Peptide-1 (GLP-1) Receptor by Peptide and Non-Peptide Ligands Clare Louise Wishart Submitted in accordance with the requirements for the degree of Doctor of Philosophy of Science University of Leeds School of Biomedical Sciences Faculty of Biological Sciences September 2013 I Intellectual Property and Publication Statements The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement. The right of Clare Louise Wishart to be identified as Author of this work has been asserted by her in accordance with the Copyright, Designs and Patents Act 1988. © 2013 The University of Leeds and Clare Louise Wishart. II Acknowledgments Firstly I would like to offer my sincerest thanks and gratitude to my supervisor, Dr. Dan Donnelly, who has been nothing but encouraging and engaging from day one. I have thoroughly enjoyed every moment of working alongside him and learning from his guidance and wisdom. My thanks go to my academic assessor Professor Paul Milner whom I have known for several years, and during my time at the University of Leeds he has offered me invaluable advice and inspiration. Additionally I would like to thank my academic project advisor Dr. Michael Harrison for his friendship, help and advice. I would like to thank Dr. Rosalind Mann and Dr. Elsayed Nasr for welcoming me into the lab as a new PhD student and sharing their experimental techniques with me, these techniques have helped me no end in my time as a research student. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

AP2A2 Antibody Cat
AP2A2 Antibody Cat. No.: 63-513 AP2A2 Antibody Formalin-fixed and paraffin-embedded human Flow cytometric analysis of HepG2 cells using AP2A2 hepatocarcinoma with AP2A2 Antibody , which was Antibody (bottom histogram) compared to a negative peroxidase-conjugated to the secondary antibody, control cell (top histogram). FITC-conjugated goat-anti- followed by DAB staining. rabbit secondary antibodies were used for the analysis. Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human This AP2A2 antibody is generated from rabbits immunized with a KLH conjugated IMMUNOGEN: synthetic peptide between 610-637 amino acids from the Central region of human AP2A2. TESTED APPLICATIONS: Flow, IHC-P, WB For WB starting dilution is: 1:1000 APPLICATIONS: For IHC-P starting dilution is: 1:10~50 For FACS starting dilution is: 1:10~50 September 25, 2021 1 https://www.prosci-inc.com/ap2a2-antibody-63-513.html PREDICTED MOLECULAR 104 kDa WEIGHT: Properties This antibody is purified through a protein A column, followed by peptide affinity PURIFICATION: purification. CLONALITY: Polyclonal ISOTYPE: Rabbit Ig CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: Supplied in PBS with 0.09% (W/V) sodium azide. CONCENTRATION: batch dependent Store at 4˚C for three months and -20˚C, stable for up to one year. As with all antibodies STORAGE CONDITIONS: care should be taken to avoid repeated freeze thaw cycles. Antibodies should not be exposed to prolonged high temperatures. Additional Info OFFICIAL SYMBOL: AP2A2 AP-2 complex subunit alpha-2, 100 kDa coated vesicle -

RNA-Binding Protein Hnrnpll Regulates Mrna Splicing and Stability During B-Cell to Plasma-Cell Differentiation
RNA-binding protein hnRNPLL regulates mRNA splicing and stability during B-cell to plasma-cell differentiation Xing Changa,b, Bin Lic, and Anjana Raoa,b,d,e,1 Divisions of aSignaling and Gene Expression and cVaccine Discovery, La Jolla Institute for Allergy and Immunology, La Jolla, CA 92037; bSanford Consortium for Regenerative Medicine, La Jolla, CA 92037; and dDepartment of Pharmacology and eMoores Cancer Center, University of California at San Diego, La Jolla, CA 92093 Contributed by Anjana Rao, December 2, 2014 (sent for review July 20, 2014) Posttranscriptional regulation is a major mechanism to rewire the RBP-binding sites, thus validating the specificity of RBP binding transcriptomes during differentiation. Heterogeneous nuclear to coprecipitating RNAs and mapping RBP-binding sites on the RNA-binding protein LL (hnRNPLL) is specifically induced in terminally validated RNAs at close to single-nucleotide resolution (8). differentiated lymphocytes, including effector T cells and plasma Heterogeneous nuclear RNA-binding proteins (hnRNPs) is cells. To study the molecular functions of hnRNPLL at a genome- the term applied to a collection of unrelated nuclear RBPs. wide level, we identified hnRNPLL RNA targets and binding sites in hnRNPLL was identified through a targeted lentiviral shRNA plasma cells through integrated Photoactivatable-Ribonucleoside- screen for regulators of CD45RA to CD45RO switching during Enhanced Cross-Linking and Immunoprecipitation (PAR-CLIP) and memory T-cell development (9) and independently through RNA sequencing. hnRNPLL preferentially recognizes CA dinucleo- two separate screens performed by different groups for exclusion tide-containing sequences in introns and 3′ untranslated regions of CD45 exon 4 in a minigene context (10) and for altered CD44 (UTRs), promotes exon inclusion or exclusion in a context-dependent and CD45R expression on T cells in N-ethyl-N-nitrosourea manner, and stabilizes mRNA when associated with 3′ UTRs. -

Ovarian Gene Expression in the Absence of FIGLA, an Oocyte
BMC Developmental Biology BioMed Central Research article Open Access Ovarian gene expression in the absence of FIGLA, an oocyte-specific transcription factor Saurabh Joshi*1, Holly Davies1, Lauren Porter Sims2, Shawn E Levy2 and Jurrien Dean1 Address: 1Laboratory of Cellular and Developmental Biology, NIDDK, National Institutes of Health, Bethesda, MD 20892, USA and 2Department of Biomedical Informatics, Vanderbilt University Medical Center, Nashville, TN 37232, USA Email: Saurabh Joshi* - [email protected]; Holly Davies - [email protected]; Lauren Porter Sims - [email protected]; Shawn E Levy - [email protected]; Jurrien Dean - [email protected] * Corresponding author Published: 13 June 2007 Received: 11 December 2006 Accepted: 13 June 2007 BMC Developmental Biology 2007, 7:67 doi:10.1186/1471-213X-7-67 This article is available from: http://www.biomedcentral.com/1471-213X/7/67 © 2007 Joshi et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Ovarian folliculogenesis in mammals is a complex process involving interactions between germ and somatic cells. Carefully orchestrated expression of transcription factors, cell adhesion molecules and growth factors are required for success. We have identified a germ-cell specific, basic helix-loop-helix transcription factor, FIGLA (Factor In the GermLine, Alpha) and demonstrated its involvement in two independent developmental processes: formation of the primordial follicle and coordinate expression of zona pellucida genes. Results: Taking advantage of Figla null mouse lines, we have used a combined approach of microarray and Serial Analysis of Gene Expression (SAGE) to identify potential downstream target genes. -
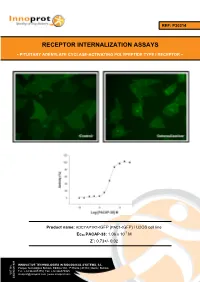
Receptor Internalization Assays
REF: P30214 RECEPTOR INTERNALIZATION ASSAYS - PITUITARY ADENYLATE CYCLASE-ACTIVATING POLYPEPTIDE TYPE I RECEPTOR - Product name: ADCYAP1R1-tGFP (PAC1-tGFP) / U2OS cell line -7 Ec50 PACAP-38: 1.06 x 10 M Z´: 0.73+/- 0.02 INNOVATIVE TECHNOLOGIES IN BIOLOGICAL SYSTEMS, S.L. Parque Tecnológico Bizkaia, Edifício 502, 1ª Planta | 48160 | Derio | Bizkaia Tel.: +34 944005355 | Fax: +34 946579925 VAT No. [email protected] | www.innoprot.com ESB95481909 Product Name: ADCYAP1R1-tGFP_U2OS Reference: P30214 Rep. Official Full Name: Pituitary adenylate cyclase- activating polypeptide type I receptor DNA Accession Number: Gene Bank AY366498 Host Cell: U2OS References: P30214: 2 vials of 3 x 106 proliferative cells P30214-DA: 1 vial of 2 x 106 division-arrested cells Storage: Liquid Nitrogen Assay Briefly description About ADCYAP1R1 Each vial of ADCYAP1R1 Internalization Assay Pituitary adenylate cyclase-activating Cell Line contains U2OS cells stably expressing polypeptide type I receptor, also known as human Pituitary adenylate cyclase-activating PAC1 is a protein that in humans is encoded by polypeptide type I receptor tagged in the N- the ADCYAP1R1 gene. ADCYAP1R1 is a terminus with tGFP protein. membrane-associated protein and shares significant homology with members of the Innoprot’s ADCYAP1R1-tGFP Internalization glucagon/secretin receptor family. This receptor Assay Cell Line has been designed to assay binds pituitary adenylate cyclase activating potential agonists/ antagonists against peptide (PACAP) mediating several biological ADCYAP1R1, modulating its activation and the activities and it is positively coupled to following redistribution process inside the cells. adenylate cyclase. This cell line will allow the image analysis of the stimuli induced by the compounds. -

Constitutive Activation of RAS/MAPK Pathway Cooperates with Trisomy 21 and Is Therapeutically Exploitable in Down Syndrome B-Cell Leukemia
Author Manuscript Published OnlineFirst on March 27, 2020; DOI: 10.1158/1078-0432.CCR-19-3519 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Constitutive activation of RAS/MAPK pathway cooperates with trisomy 21 and is therapeutically exploitable in Down syndrome B-cell Leukemia Anouchka P. Laurent1,2, Aurélie Siret1, Cathy Ignacimouttou1, Kunjal Panchal3, M’Boyba K. Diop4, Silvia Jenny5, Yi-Chien Tsai5, Damien Ross-Weil1, Zakia Aid1, Naïs Prade6, Stéphanie Lagarde6, Damien Plassard7, Gaelle Pierron8, Estelle Daudigeos-Dubus4, Yann Lecluse4, Nathalie Droin1, Beat Bornhauser5, Laurence C. Cheung3,9, John D. Crispino10, Muriel Gaudry1, Olivier A. Bernard1, Elizabeth Macintyre11, Carole Barin Bonnigal12, Rishi S. Kotecha3,9,13, Birgit Geoerger4, Paola Ballerini14, Jean-Pierre Bourquin5, Eric Delabesse6, Thomas Mercher1,15 and Sébastien Malinge1,3 1INSERM U1170, Gustave Roussy Institute, Université Paris Saclay, Villejuif, France 2Université Paris Diderot, Paris, France 3Telethon Kids Cancer Centre, Telethon Kids Institute, University of Western Australia, Perth, Australia 4Gustave Roussy Institute Cancer Campus, Department of Pediatric and Adolescent Oncology, INSERM U1015, Equipe Labellisée Ligue Nationale contre le Cancer, Université Paris-Saclay, Villejuif, France 5Department of Pediatric Oncology, Children’s Research Centre, University Children’s Hospital Zurich, Zurich, Switzerland 6Centre of Research on Cancer of Toulouse (CRCT), CHU Toulouse, Université Toulouse III, Toulouse, France 7IGBMC, Plateforme GenomEast, UMR7104 CNRS, Ilkirch, France 8Service de Génétique, Institut Curie, Paris, France 9School of Pharmacy and Biomedical Sciences, Curtin University, Bentley, Australia 10Division of Hematology/Oncology, Northwestern University, Chicago, USA 11Hematology, Université de Paris, Institut Necker-Enfants Malades and Assistance Publique – Hopitaux de Paris, Paris, France 12Centre Hospitalier Universitaire de Tours, Tours, France 1 Downloaded from clincancerres.aacrjournals.org on September 30, 2021. -

Protein Interaction Network of Alternatively Spliced Isoforms from Brain Links Genetic Risk Factors for Autism
ARTICLE Received 24 Aug 2013 | Accepted 14 Mar 2014 | Published 11 Apr 2014 DOI: 10.1038/ncomms4650 OPEN Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism Roser Corominas1,*, Xinping Yang2,3,*, Guan Ning Lin1,*, Shuli Kang1,*, Yun Shen2,3, Lila Ghamsari2,3,w, Martin Broly2,3, Maria Rodriguez2,3, Stanley Tam2,3, Shelly A. Trigg2,3,w, Changyu Fan2,3, Song Yi2,3, Murat Tasan4, Irma Lemmens5, Xingyan Kuang6, Nan Zhao6, Dheeraj Malhotra7, Jacob J. Michaelson7,w, Vladimir Vacic8, Michael A. Calderwood2,3, Frederick P. Roth2,3,4, Jan Tavernier5, Steve Horvath9, Kourosh Salehi-Ashtiani2,3,w, Dmitry Korkin6, Jonathan Sebat7, David E. Hill2,3, Tong Hao2,3, Marc Vidal2,3 & Lilia M. Iakoucheva1 Increased risk for autism spectrum disorders (ASD) is attributed to hundreds of genetic loci. The convergence of ASD variants have been investigated using various approaches, including protein interactions extracted from the published literature. However, these datasets are frequently incomplete, carry biases and are limited to interactions of a single splicing isoform, which may not be expressed in the disease-relevant tissue. Here we introduce a new interactome mapping approach by experimentally identifying interactions between brain-expressed alternatively spliced variants of ASD risk factors. The Autism Spliceform Interaction Network reveals that almost half of the detected interactions and about 30% of the newly identified interacting partners represent contribution from splicing variants, emphasizing the importance of isoform networks. Isoform interactions greatly contribute to establishing direct physical connections between proteins from the de novo autism CNVs. Our findings demonstrate the critical role of spliceform networks for translating genetic knowledge into a better understanding of human diseases. -

Epithelial Delamination Is Protective During Pharmaceutical-Induced Enteropathy
Epithelial delamination is protective during pharmaceutical-induced enteropathy Scott T. Espenschieda, Mark R. Cronana, Molly A. Mattya, Olaf Muellera, Matthew R. Redinbob,c,d, David M. Tobina,e,f, and John F. Rawlsa,e,1 aDepartment of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC 27710; bDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599; cDepartment of Biochemistry, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599; dDepartment of Microbiology and Immunology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, NC 27599; eDepartment of Medicine, Duke University School of Medicine, Durham, NC 27710; and fDepartment of Immunology, Duke University School of Medicine, Durham, NC 27710 Edited by Dennis L. Kasper, Harvard Medical School, Boston, MA, and approved July 15, 2019 (received for review February 12, 2019) Intestinal epithelial cell (IEC) shedding is a fundamental response to in mediating intestinal responses to injury remains poorly un- intestinal damage, yet underlying mechanisms and functions have derstood for most xenobiotics. been difficult to define. Here we model chronic intestinal damage in Gastrointestinal pathology is common in people using phar- zebrafish larvae using the nonsteroidal antiinflammatory drug maceuticals, including nonsteroidal antiinflammatory drugs (NSAID) Glafenine. Glafenine induced the unfolded protein response (NSAIDs) (11). While gastric ulceration has historically been a (UPR) and inflammatory pathways in IECs, leading to delamination. defining clinical presentation of NSAID-induced enteropathy, Glafenine-induced inflammation was augmented by microbial colo- small intestinal pathology has also been observed, although the nizationandassociatedwithchanges in intestinal and environmental incidence may be underreported due to diagnostic limitations microbiotas. -

Global Analysis of Protein Folding Thermodynamics for Disease State Characterization
Global Analysis of Protein Folding Thermodynamics for Disease State Characterization and Biomarker Discovery by Jagat Adhikari Department of Biochemistry Duke University Date:_______________________ Approved: ___________________________ Michael C. Fitzgerald, Supervisor ___________________________ Kenneth Kreuzer ___________________________ Terrence G. Oas ___________________________ Jiyong Hong ___________________________ Seok-Yong Lee Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biochemistry in the Graduate School of Duke University 2015 ABSTRACT Global Analysis of Protein Folding Thermodynamics for Disease State Characterization and Biomarker Discovery by Jagat Adhikari Department of Biochemistry Duke University Date:_______________________ Approved: ___________________________ Michael C. Fitzgerald, Supervisor ___________________________ Kenneth Kreuzer ___________________________ Terrence G. Oas ___________________________ Jiyong Hong ___________________________ Seok-Yong Lee An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Biochemistry in the Graduate School of Duke University 2015 Copyright by Jagat Adhikari 2015 Abstract Protein biomarkers can facilitate the diagnosis of many diseases such as cancer and they can be important for the development of effective therapeutic interventions. Current large-scale biomarker discovery and disease state characterization -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human.