The Mechanism and Kinetics of Methyl Isobutyl Ketone Synthesis from Acetone Over Ion-Exchanged Hydroxyapatite ⇑ Christopher R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alcohols Combined 1405
ALCOHOLS COMBINED 1405 Formulas: Table 1 MW: Table 1 CAS: Table 2 RTECS: Table 2 METHOD: 1405, Issue 1 EVALUATION: PARTIAL Issue 1: 15 March 2003 OSHA : Table 2 PROPERTIES: Table 1 NIOSH: Table 2 ACGIH: Table 2 COMPOUNDS: (1) n-butyl alcohol (4) n-propyl alcohol (7) cyclohexanol (2) sec-butyl alcohol (5) allyl alcohol (8) isoamyl alcohol (3) isobutyl alcohol (6) diacetone alcohol (9) methyl isobutyl carbinol SYNONYMS: See Table 3. SAMPLING MEASUREMENT SAMPLER: SOLID SORBENT TUBE TECHNIQUE: GAS CHROMATOGRAPHY, FID (Coconut shell charcoal, 100 mg/50 mg) ANALYTE: Compounds above FLOW RATE: 0.01 to 0.2 L/min DESORPTION: 1 mL 5% 2-propanol in CS2 Compounds: (1-3 ) (4-9) VOL-MIN: 2 L 1 L INJECTION -MAX: 10 L 10 L VOLUME: 1 µL SHIPMENT: Routine TEMPERATURE -INJECTION: 220 °C SAMPLE -DETECTOR: 250 - 300 °C STABILITY: See Evaluation of Method. -COLUMN: 35 °C (7 minutes), to 60 °C at 5 °C/minute, hold 5 minutes, up to BLANKS: 2 to 10 field blanks per set 120 °C at 10 °C /minute, hold 3 minutes. CARRIER GAS: He, 4 mL/min ACCURACY COLUMN: Capillary, fused silica, 30 m x 0.32-mm RANGE STUDIED: Not studied [1, 2]. ID; 0.5 µm film polyethylene glycol, DB- wax or equivalent BIAS: Not determined CALIBRATION: Solutions of analyte in eluent (internal OVERALL standard optional) PRECISION (Ö ): Not determined rT RANGE: See EVALUATION OF METHOD. ACCURACY: Not determined ESTIMATED LOD: 1 µg each analyte per sample PRECISION: See EVALUATION OF METHOD. APPLICABILITY: This method may be used to determine two or more of the specified analytes simultaneously. -

Supporting Information
Supporting Information Section 1 Components of DCM based coating strippers Table 1. Composition of Klean Strip Premium. CAS # Components Concentration 75-09-2 Dichloromethane 70.0-95.0% 67-56-1 Methanol < 5.0% 127087-87-0 Poly(oxy-1,2-ethandiyl) < 5.0% 124-38-9 Carbon dioxide < 5.0% Table 2. Composition of Klean Strip X. CAS # Components Concentration 75-09-2 Dichloromethane 30.0 – 40.0% 67-56-1 Methanol 15.0 – 26.0% 67-64-1 Acetone < 10.0% 1330-20-7 Xylene < 10.0% 108-88-3 Toluene < 10.0% 100-41-4 Ethylbenzene < 5.0% 64-17-5 Ethyl alcohol < 5.0% 67-63-0 Isopropyl alcohol < 5.0% Section 2 Sample preparation for the dwell time test As Figure S1 shows, a gasket was pasted on the conformal coating surface and a sheet of parafilm attached to the back of the printed circuit board to avoid solvent leakage dur- ing the dwell test. Figure 1. Sample preparation for the dwell time test. Section 3 Thickness measurement of coated PCBs Materials and Equipment Printed circuit boards (PCB), acrylic conformal coating, tape, Dektak stylus profiler (Bruker, Arizona, USA). Methods A piece of tape was attached on a PCB before coating. The coating was applied on the PCB using the same method as dip coating in the dwell time test. The PCB was sta- tioned and dried at room temperature for over 24 hours. The tape was then torn out to create a coating step. The PCB was fixed on the detection table using tapes. The stylus scanned from sub- strate to coated area. -

Silfort* SHC1200 Silfort* SHC1200
Technical Data Sheet SilFORT* SHC1200 SilFORT* SHC1200 Description SilFORT SHC1200 Hard Coat SHC1200 hard coat has been found to yield a clear mar-resistant film when applied to a suitably prepared plastic substrate. It can be applied by flow, dip or spray coating. SilFORT SHP401 Primer SHP401 air-dried primer is used as an adhesion promoter for SHC1200 hard coat on polycarbonate resin. It can be applied by flow, dip or spray coating. Key Features and Benefits Fast cure Abrasion resistance Scratch resistance Good clarity Solvent/chemical resistance SHP401 Primer No thermal cure required Improves coating adhesion Improves water resistance Page 1 of 6 *SilFORT ist ein Markenname der Momentive Performance Materials Inc. SilFORT* SHC1200 Improves ultraviolet resistance SHP401 Primer/SHC1200 Hard Coat on polycarbonate (2 - 4 micron Topcoat Thickness) Cured Film Properties Film Thickness, slow dip coat, 10-18 cm/min 2 – 4 micron withdrawal,18-20% solids at 22C Taber Abrasion, 500 cycles 500G on primed < 6.0 % Haze measured per polycarbonate (CS10F wheel) ASTM D1003. * Index of Refraction 1.4 *Humidity during coating and testing will affect final values. Typical Physical Properties Property SHC1200 Hard Coat SHP401 Primer Solids Content, % 20 ± 1 2.1 ± 0.2 Methanol, Isobutanol, 1-Methoxy-2-propanol, Diacetone Solvent Isopropanol Alcohol Flash Point, PMCC 19.4C 36.1C Density, g/cm3 0.911 0.959 pH 7.3 ± 0.2 - Viscosity, cstk @ 25°C 20 ± 3 4 - 7 Dry Film Thickness, 2.75 - 4.5 0.5 micron VOC, g/l 710 937 Patent Status Nothing contained herein shall be construed to imply the nonexistence of any relevant patents or to constitute the permission, inducement or recommendation to practice any invention covered by any patent, without authority from the owner of the patent. -
Methanol and Acetone Is in Organic Synthesis
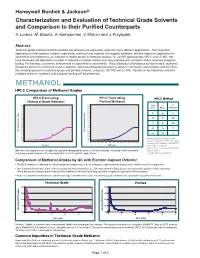
Honeywell Burdick & Jackson® Characterization and Evaluation of Technical Grade Solvents and Comparison to their Purified Counterparts S. Lorenz, M. Bosma, A. Kemperman, V. Mohan and J. Przybytek Abstract: Technical grade solvents and their purified counterparts are separately useful for many different applications. One important application for the common solvents acetonitrile, methanol and acetone is in organic synthesis. Another important application for acetonitrile and methanol is as a diluent or mobile phase in chemical analysis, ie: UV-VIS spectroscopy, HPLC and LC-MS. We have observed and identified a number of impurities in these solvents that may interfere with synthesis and/or accurate analytical testing. For instance, a common contaminant in acetonitrile is acrylonitrile. Trace aldehydes and ketones can be found in methanol. Diacetone alcohol is commonly found in acetone. We have characterized impurity levels in commonly used solvents and will show the variability present in technical grade and purified solvents, using GC, GC-MS and LC-MS. Results on the impurities and their probable effect on synthesis and analytical testing will be presented. METHANOL HPLC Comparison of Methanol Grades HPLC Trace using HPLC Trace using HPLC Method Technical Grade Methanol Purified Methanol 0.200 0.200 Time % % 0.190 0.190 0.180 0.180 (min) Water Methanol 0.170 0.170 0.160 0.160 0.150 0.150 0 95 5 0.140 AU) 0.140 AU) ( ( 0.130 0.130 e e 20 0 100 c 0.120 c 0.120 n 0.110 n 0.110 a a b 0.100 b 0.100 r r 25 0 100 o 0.090 o 0.090 s s 0.080 0.080 -

US EPA, Inert (Other) Pesticide Ingredients in Pesticide Products
Inert Ingredients ordered by CAS Number Updated August 2004 CAS PREFIX NAME List No. 50-21-5 Lactic acid 4B 50-70-4 Sorbitol 4A 50-81-7 L- Ascorbic acid 4A 50-99-7 Dextrose 4A 51-03-6 Piperonyl butoxide 3 51-05-8 Procaine hydrochloride 3 51-55-8 Atropine 3 52-51-7 2- Bromo-2-nitro-propane-1,3-dio 3 54-21-7 Sodium salicylate 3 56-81-5 Glycerol (glycerin) 1,2,3 propanetriol 4A 56-86-0 L- Glutamic acid 3 56-95-1 Chlorhexidine diacetate 3 57-10-3 Hexadecanoic acid 4A 57-11-4 Stearic acid 4A 57-13-6 Urea 4A 57-48-7 D- Fructose 4B 57-50-1 Sugar 4A 57-55-6 Propylene glycol 4B 57-88-5 (3.beta.)- Cholest-5-en-3-ol 4B 58-08-2 1H- Purine-2,6-dione, 3,7-dihydro-1,3,7-trimethyl- 4B 58-56-0 Thiamine mononitrate 4B 58-85-5 Biotin 3 58-86-6 D- Xylose 4B 58-95-7 Vitamin E acetate 3 59-30-3 Folic acid 4B 59-40-5 N-(2- Quinoxalinyl)sulfanilide 3 59-67-6 Nicotinic acid 3 60-00-4 Ethylenediaminetetraacetic acid (EDTA) 4B 60-12-8 Benzeneethanol 3 60-29-7 Ethane, 1,1'-oxybis- 3 60-33-3 Linoleic acid 3 61-73-4 C.I. Basic Blue 9 3 62-33-9 Ethylenediaminetetraacetic acid (EDTA), calcium4B 62-54-4 Acetic acid, calcium salt 4A 63-42-3 D-(+)-Lactose 4A 63-68-3 L- Methionine 4B 64-02-8 Ethylenediaminetetraacetic acid (EDTA), tetraso4B 64-17-5 Ethanol 4B 64-18-6 Formic acid 3 64-19-7 Acetic acid 4B 64-86-8 Colchicine 3 65-85-0 Benzoic acid 4B 66-71-7 1,10- Phenanthroline 3 67-03-8 Thiamin hydrochloride 3 67-43-6 1,1,4,7,7- Diethylenetriaminepentaacetic acid 3 67-48-1 Choline chloride 4B 67-56-1 Methyl alcohol 3 67-63-0 2- Propanol 4B 67-64-1 Acetone 3 67-68-5 Dimethyl -

Safety Assessment of Diacetone Alcohol As Used in Cosmetics
Safety Assessment of Diacetone Alcohol as Used in Cosmetics Status: Draft Tentative Report for Panel Review Release Date: February 16, 2021 Panel Meeting Date: March 11 – 12, 2021 The Expert Panel for Cosmetic Ingredient Safety members are: Chair, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; David E. Cohen, M.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; Lisa A. Peterson, Ph.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. Previous Panel member involved in this assessment: James G. Marks, Jr., M.D. The Cosmetic Ingredient Review (CIR) Executive Director is Bart Heldreth, Ph.D. This safety assessment was prepared by Priya Cherian, Scientific Analyst/Writer, CIR. © Cosmetic Ingredient Review 1620 L Street, NW, Suite 1200 ♢ Washington, DC 20036-4702 ♢ ph 202.331.0651 ♢ fax 202.331.0088 ♢ [email protected] Distributed for Comment Only -- Do Not Cite or Quote Commitment & Credibility since 1976 Memorandum To: Expert Panel for Cosmetic Ingredient Safety Members and Liaisons From: Priya Cherian, Scientific Analyst/Writer, CIR Date: February 16, 2021 Subject: Safety Assessment of Diacetone Alcohol as Used in Cosmetics Enclosed is the Draft Tentative Report of the Safety Assessment of Diacetone Alcohol as Used in Cosmetics (diacet032021rep). At the September 2020 meeting, the Expert Panel for Cosmetic Ingredient Review Safety (Panel) issued an Insufficient Data Announcement (IDA) for this ingredient. In order to come to a conclusion of safety, the Panel requested impurities data. Since the previous review of this report, no new data have been received. -

Trace Analysis of Mesityl Oxide and Diacetone Alcohol in Pharmaceuticals by Capillary Gas Chromatography with Flame Ionization Detection
id22818046 pdfMachine by Broadgun Software - a great PDF writer! - a great PDF creator! - http://www.pdfmachine.com http://www.broadgun.com AASeptemnnbeaar 2ll0y0y9ttiiccaall Volume 8 Issue 3 An Indian Journal Trade Science Inc. CCHHEEMMIISSTTRRYY Full Paper ACAIJ, 8(3) 2009 [346-349] Trace analysis of mesityl oxide and diacetone alcohol in pharmaceuticals by capillary gas chromatography with flame ionization detection K.Rama Seshaiah*1,2, V.Krishna Reddy1, Ch.Krishnaiah1, VVNKV Prasadaraju1, K.Mukkanti2, V.Ranga Reddy1 ’ 1Dr.Reddy s Laboratories Ltd. Active pharmaceutical ingredients, IPDO, Bachupally, Hyderabad-500072, A.P, (INDIA) 2Department of chemisty, J.N.T.University, Kukatpally, Hyderabad-500072, A.P, (INDIA) E-mail: [email protected] Received: 26th May, 2009 ; Accepted: 5th June, 2009 ABSTRACT KEYWORDS A capillary gas chromatographic method using flame ionization detection Pharmaceutical analysis; was developed and validated for the trace analysis (ppm level) of mesityl Mesityl oxide and Diacetone oxide and diacetone alcohol in pharmaceutical drug substance. The method alcohol. utilizes a megabore capillary column with bonded and crosslinked polyeth- ylene glycol stationary phase. A dissolve-and-injection approach was adopted for sample introduction in a split mode (1:1). Water is used as ppm and limit of sample solvent. A limit of detection of about 1.5 and 2 ppm were achieved for the Mesityl oxide and quantitation of about 6 and 10 Diacetone alcohol in drug substance samples. The method optimization and validation are also discussed in this paper. 2009 Trade Science Inc. - INDIA INTRODUCTION ods that are sensitive enough and meet all the regula- tory requirements. Recently, the potential health hazards of trace The pure mesityl oxide and diacetone alcohol are amounts of mesityl oxide and diacetone alcohol in phar- liquids at ambient temperature with a boiling point °C and 170°C respectively. -

Final Report of the Addendum to The
International Journal of Toxicology, 27(Suppl. 2f53—69, 2008 Copyright © American College of Toxicology ISSN: 1091-5818 print! 1092-874X online 001: 10.1080/10915810802244504 Final Report of the Addendum to the Safety Assessment of n-Butyl Alcohol as Used in Cosmetics’ n-Butyl Alcohol is a primary aliphatic alcohol historically used in care cosmetic products, but new concentration as a solvent nail INTRODUCTION of use data indicate that it also is being used at low concentrations in eye makeup, personal hygiene, and shaving cosmetic products. The Cosmetic Ingredient Review (CIR) evaluated the safety it-Butyl Alcohol has been generally recognized as safe for use as a of n-Butyl Alcohol (n-BuOH) in 1987, finding it safe in the flavoring substance in food and appears on the 1982 Food and Drug practices of use and concentration in nail products (Elder 1987). list of inactive ingredients for approved pre Administration (FDA) This original safety assessment was specific in that the conclu scription drug products. n-Butyl Alcohol can be absorbed through regards the use of n-Butyl Alcohol in the skin, tangs, and gastrointestinal tract. n-Butyl Alcohol may be sion was issued only as formed by hydrolysis of butyl acetate in the blood, but is rapidly nail products. oxidized. The single oral dose ED50 of n-Butyl Alcohol for rats was Recently, CIR undertook a re-review of this ingredient to de 0.79 to 4.36 g/kg. The dermal ED50 for rabbits was 4.2 g/kg. Inhala termine what additional data relevant to the safety of n-Butyl Al humans demonstrate sensory irritation of tion toxicity studies in 3. -

These Two Workbooks Are Provided by As a Convenient Introduc
These two workbooks are provided by www.hansen-solubility.com as a convenient introduc They are Copyright © 2013 Prof Steven Abbott If you find bugs/issues or would like extra functionality, please email Steven Abbott [email protected] ction to some of the basic HSP methods HSP Sphere dD dP dH R Good 11 18.4 9.7 8.0 7.1 Bad 11 Test Value 16 7 8 Total 22 Delta 2.4 11.4 10.4 Distance 5.5 RED 0.77 Solvents dD dP dH MVol Score Distance Acetone 15.5 10.4 7 73.8 1 5.915773 Acetonitrile 15.3 18 6.1 52.9 0 10.52754 n-Amyl Acetate 15.8 3.3 6.1 148 0 n-Amyl Alcohol 15.9 5.9 13.9 108.6 0 Benzene 18.4 0 2 52.9 0 11.38507 Benzyl Alcohol 18.4 6.3 13.7 103.8 0 Benzyl Benzoate 20 5.1 5.2 190.3 0 1-Butanol 16 5.7 15.8 92 0 2-Butanol 15.8 5.7 14.5 92 0 n-Butyl Acetate 15.8 3.7 6.3 132.6 0 t-Butyl Acetate 15 3.7 6 134.8 0 t-Butyl Alcohol 15.2 5.1 14.7 96 0 Butyl Benzoate 18.3 5.6 5.5 178.1 0 Butyl Diglycol Acetate 16 4.1 8.2 208.2 0 Butyl Glycol Acetate 15.3 7.5 6.8 171.2 0 n-Butyl Propionate 15.7 5.5 5.9 149.3 0 Caprolactone (Epsilon) 19.7 15 7.4 110.8 0 Chloroform 17.8 3.1 5.7 80.5 1 7.075453 m-Cresol 18.5 6.5 13.7 105 1 6.563973 Cyclohexane 16.8 0 0.2 108.9 0 12.82752 Cyclohexanol 17.4 4.1 13.5 105.7 0 Cyclohexanone 17.8 8.4 5.1 104.2 0 Di-isoButyl Ketone 16 3.7 4.1 177.4 0 Diacetone Alcohol 15.8 8.2 10.8 124.3 0 Diethyl Ether 14.5 2.9 4.6 104.7 0 10.87429 Diethylene Glycol Monobut 16 7 10.6 170.4 0 Dimethyl Cyclohexane 16.1 0 1.1 140 0 Dimethyl Sulfoxide (DMSO) 18.4 16.4 10.2 71.3 1 7.066692 1,4-Dioxane 17.5 1.8 9 85.7 0 8.160898 1,3-Dioxolane -

Chemical Compatibility Chart
Chemical Compatibility Chart 1 Inorganic Acids 1 2 Organic acids X 2 3 Caustics X X 3 4 Amines & Alkanolamines X X 4 5 Halogenated Compounds X X X 5 6 Alcohols, Glycols & Glycol Ethers X 6 7 Aldehydes X X X X X 7 8 Ketone X X X X 8 9 Saturated Hydrocarbons 9 10 Aromatic Hydrocarbons X 10 11 Olefins X X 11 12 Petrolum Oils 12 13 Esters X X X 13 14 Monomers & Polymerizable Esters X X X X X X 14 15 Phenols X X X X 15 16 Alkylene Oxides X X X X X X X X 16 17 Cyanohydrins X X X X X X X 17 18 Nitriles X X X X X 18 19 Ammonia X X X X X X X X X 19 20 Halogens X X X X X X X X X X X X 20 21 Ethers X X X 21 22 Phosphorus, Elemental X X X X 22 23 Sulfur, Molten X X X X X X 23 24 Acid Anhydrides X X X X X X X X X X 24 X Represents Unsafe Combinations Represents Safe Combinations Group 1: Inorganic Acids Dichloropropane Chlorosulfonic acid Dichloropropene Hydrochloric acid (aqueous) Ethyl chloride Hydrofluoric acid (aqueous) Ethylene dibromide Hydrogen chloride (anhydrous) Ethylene dichloride Hydrogen fluoride (anhydrous) Methyl bromide Nitric acid Methyl chloride Oleum Methylene chloride Phosphoric acid Monochlorodifluoromethane Sulfuric acid Perchloroethylene Propylene dichloride Group 2: Organic Acids 1,2,4-Trichlorobenzene Acetic acid 1,1,1-Trichloroethane Butyric acid (n-) Trichloroethylene Formic acid Trichlorofluoromethane Propionic acid Rosin Oil Group 6: Alcohols, Glycols and Glycol Ethers Tall oil Allyl alcohol Amyl alcohol Group 3: Caustics 1,4-Butanediol Caustic potash solution Butyl alcohol (iso, n, sec, tert) Caustic soda solution Butylene -

Ep 0095783 A2
Europaisch.es Patentamt European Patent Office ® Publication number: 0 095 783 A2 Office europeen des brevets ® EUROPEAN PATENT APPLICATION © Application number: 83105450.7 ® Int. CI.3: C 07 C 45/74, C 07 C 49/203, C 07 C B 01 J ^ 49/603, 29/04, @ Dateof filing: 01.06.83 B 01 J 21/16 (§) Priority: 02.06.82 US 384212 @ Applicant: UNION CARBIDE CORPORATION, Old Ridgebury Road, Danbury Connecticut 06817 (US) @@on ~ ... ,.... ,... nun on ^(7?) Inventor: Reichle, Walter Thomas, 158 Mountain Avenue, j^J/^ Warren New Jersey 07060 (US) @ Representative : Wuesthoff , Franz, Dr.-lng. et al, Patentanwalte Wuesthoff -v. Pechmann-Behrens-Goetz @ Designated Contracting States : DE FR GB Schweigerstrasse 2, D-8000 Miinchen 90 (DE) @ Catalyzed aldol condensations. Heat treated anionic clay mineral is an improved catalyst for the conversion of acetone to mesityl oxide and isophorone as well as for the aldol condensation of other carbonyl-contain- ing compounds. BACKGROUND OF THE INVENTION This invention pertains to aldol condensations catalyzed by heat-treated synthetic anionic clay minerals ana more particularly to the use of these catalysts ror the conversion of acetone to mesityl oxide and isophorone. The alaol condensation of active hydrogen-containing organic carbonyl compounds has found wide use in the chemical industry for the synthesis of a myriad of organic compounds. The earliest catalysts used for this conaensation reaction were bases, such as, alkali metal hydroxides which have been used for the production of 2-ethylhexanediol-l, 3, 2-ethylhexanol-l, diacetone alcohol, isopnorone, mesityl oxide, methylisoamyl ketone, and methylisobutyl ketone. A variety of methods has been disclosed in the literature for converting, for example, acetone by aldol condensation into a wide spectrum of products particularly isophorone and mesityl oxide which are used in industrial solvents and as chemical intermediates for resins, dyes, intersecticiaes. -

Diacetone Alcohol Decomposition and Benzaldehyde Cannizzaro Reaction
Reaction Kinetics, Mechanisms and Catalysis https://doi.org/10.1007/s11144-018-1492-z Diacetone alcohol decomposition and benzaldehyde Cannizzaro reaction as test reactions for the basic strength measurements of alumina, magnesia, Amberlyst type resins (A‑15, XN 1010, A‑26, A‑21), Nafon NR 50 and solid sulfuric acid Marek Marczewski1 · Yuliya Kavalchuk1 · Urszula Ulkowska1 · Marek Gliński1 · Osazuwa Osawaru2 Received: 15 May 2018 / Accepted: 1 November 2018 © The Author(s) 2018 Abstract The decomposition of diacetone alcohol to acetone proceeding at 303 K as well as the Cannizzaro transformation of benzaldehyde at 323 K leading to benzyl alco- hol, can be regarded as useful test reactions in investigating the basic properties of solid catalysts. The minimum basic strength H_min needed to initiate the conver- sion of diacetone alcohol is within the range of 11.8–13.3, and for benzaldehyde transformation the value is 15.4. The above mentioned test reactions were used to study the basic strength of diferent solid acid–base catalysts. The conclusions can be presented as follows: (a) solid NaOH and Amberlyst type resin A-26 exhibit basic strength of H_ ≥ 15.4 respectively, the basic centers of A-21 resin possess lower strength 11.8–13.3 ≤ H_ < 15.4; (b) Amberlyst-15, Amberlyst XN 1010, Nafon NR 50 resins and solid sulfuric acid (SSA) possess basic strength of H_ < 11.8–13.3; (c) the basic strength of alumina and magnesia is 11.8–13.3 ≤ H_ < 15.4 whether they have been calcined at 753 K or not. Keywords Basicity scale · Solid bases · Test reactions · Amberlyst resins · Nafon · Alumina · Magnesia · Solid sulfuric acid * Marek Marczewski [email protected] 1 Chemistry Department, Warsaw University of Technology, Noakowskiego 3, 00‑662 Warsaw, Poland 2 Industrial Chemistry Research Institute, Rydygiera 8, 01‑793 Warsaw, Poland Vol.:(0123456789)1 3 Reaction Kinetics, Mechanisms and Catalysis Introduction The nature and activity of basic catalysts continues to be the basis of many reactions, which have found applications in industrial processes.