Methyl Isobutyl Ketone; Steven Risotto; Roc; Sept. 20, 2013
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alcohols Combined 1405
ALCOHOLS COMBINED 1405 Formulas: Table 1 MW: Table 1 CAS: Table 2 RTECS: Table 2 METHOD: 1405, Issue 1 EVALUATION: PARTIAL Issue 1: 15 March 2003 OSHA : Table 2 PROPERTIES: Table 1 NIOSH: Table 2 ACGIH: Table 2 COMPOUNDS: (1) n-butyl alcohol (4) n-propyl alcohol (7) cyclohexanol (2) sec-butyl alcohol (5) allyl alcohol (8) isoamyl alcohol (3) isobutyl alcohol (6) diacetone alcohol (9) methyl isobutyl carbinol SYNONYMS: See Table 3. SAMPLING MEASUREMENT SAMPLER: SOLID SORBENT TUBE TECHNIQUE: GAS CHROMATOGRAPHY, FID (Coconut shell charcoal, 100 mg/50 mg) ANALYTE: Compounds above FLOW RATE: 0.01 to 0.2 L/min DESORPTION: 1 mL 5% 2-propanol in CS2 Compounds: (1-3 ) (4-9) VOL-MIN: 2 L 1 L INJECTION -MAX: 10 L 10 L VOLUME: 1 µL SHIPMENT: Routine TEMPERATURE -INJECTION: 220 °C SAMPLE -DETECTOR: 250 - 300 °C STABILITY: See Evaluation of Method. -COLUMN: 35 °C (7 minutes), to 60 °C at 5 °C/minute, hold 5 minutes, up to BLANKS: 2 to 10 field blanks per set 120 °C at 10 °C /minute, hold 3 minutes. CARRIER GAS: He, 4 mL/min ACCURACY COLUMN: Capillary, fused silica, 30 m x 0.32-mm RANGE STUDIED: Not studied [1, 2]. ID; 0.5 µm film polyethylene glycol, DB- wax or equivalent BIAS: Not determined CALIBRATION: Solutions of analyte in eluent (internal OVERALL standard optional) PRECISION (Ö ): Not determined rT RANGE: See EVALUATION OF METHOD. ACCURACY: Not determined ESTIMATED LOD: 1 µg each analyte per sample PRECISION: See EVALUATION OF METHOD. APPLICABILITY: This method may be used to determine two or more of the specified analytes simultaneously. -

Toxicological Profile for 2-Butanone Released for Public Comment in May 2019
Toxicological Profile for 2-Butanone October 2020 2-BUTANONE ii DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. 2-BUTANONE iii FOREWORD This toxicological profile is prepared in accordance with guidelines* developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. The adequacy of information to determine a substance's -

Supporting Information
Supporting Information Section 1 Components of DCM based coating strippers Table 1. Composition of Klean Strip Premium. CAS # Components Concentration 75-09-2 Dichloromethane 70.0-95.0% 67-56-1 Methanol < 5.0% 127087-87-0 Poly(oxy-1,2-ethandiyl) < 5.0% 124-38-9 Carbon dioxide < 5.0% Table 2. Composition of Klean Strip X. CAS # Components Concentration 75-09-2 Dichloromethane 30.0 – 40.0% 67-56-1 Methanol 15.0 – 26.0% 67-64-1 Acetone < 10.0% 1330-20-7 Xylene < 10.0% 108-88-3 Toluene < 10.0% 100-41-4 Ethylbenzene < 5.0% 64-17-5 Ethyl alcohol < 5.0% 67-63-0 Isopropyl alcohol < 5.0% Section 2 Sample preparation for the dwell time test As Figure S1 shows, a gasket was pasted on the conformal coating surface and a sheet of parafilm attached to the back of the printed circuit board to avoid solvent leakage dur- ing the dwell test. Figure 1. Sample preparation for the dwell time test. Section 3 Thickness measurement of coated PCBs Materials and Equipment Printed circuit boards (PCB), acrylic conformal coating, tape, Dektak stylus profiler (Bruker, Arizona, USA). Methods A piece of tape was attached on a PCB before coating. The coating was applied on the PCB using the same method as dip coating in the dwell time test. The PCB was sta- tioned and dried at room temperature for over 24 hours. The tape was then torn out to create a coating step. The PCB was fixed on the detection table using tapes. The stylus scanned from sub- strate to coated area. -

The Mechanism and Kinetics of Methyl Isobutyl Ketone Synthesis from Acetone Over Ion- Exchanged Hydroxyapatite
Lawrence Berkeley National Laboratory Recent Work Title The mechanism and kinetics of methyl isobutyl ketone synthesis from acetone over ion- exchanged hydroxyapatite Permalink https://escholarship.org/uc/item/4hq4p42d Authors Ho, CR Zheng, S Shylesh, S et al. Publication Date 2018-09-01 DOI 10.1016/j.jcat.2018.07.005 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Journal of Catalysis 365 (2018) 174–183 Contents lists available at ScienceDirect Journal of Catalysis journal homepage: www.elsevier.com/locate/jcat The mechanism and kinetics of methyl isobutyl ketone synthesis from acetone over ion-exchanged hydroxyapatite ⇑ Christopher R. Ho a,b, Steven Zheng a, Sankaranarayanapillai Shylesh a,b, Alexis T. Bell a,b, a Department of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720-1462, United States b Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, United States article info abstract Article history: The synthesis of methyl isobutyl ketone (MIBK) can be carried out by the condensation of acetone in the Received 9 May 2018 presence of hydrogen over a supported metal catalyst. Previous studies have shown that hydroxyapatite Revised 26 June 2018 is an excellent catalyst for condensation reactions. The present investigation was undertaken in order to Accepted 2 July 2018 elucidate the reaction mechanism and site requirements for acetone coupling to MIBK over a physical Available online 18 July 2018 mixture of hydroxyapatite and Pd/SiO2. The reaction is found to proceed by consecutive aldol addition to form diacetone alcohol (DAA), dehydration of DAA to mesityl oxide (MO), and hydrogenation of MO Keywords: to MIBK. -

Methyl Isobutyl Ketone
Issued: Data Sheet 19-Sep-2005 Product Name Methyl isobutyl ketone Product Code S1215 Asia Pacific Product Category Ketones CAS Registry 108-10-1 Number EINECS Number 203-550-1 Alternate Name 2-methyl-2-pentanone, hexanone, MIBK Description Methyl isobutyl ketone, MIBK, a medium boiling ketone is a stable water-white liquid. Like acetone and MEK, it displays strong solvent power for cellulose esters vinyl polymers and copolymers, and most natural and synthetic resins. MIBK is a medium evaporating solvent with excellent solvency characteristics and with a high tolerance for hydrocarbon diluents. Sales Property Unit Min Max Method Specification Purity % m/m 99.5 ASTM D3329 Water % m/m 0.05 ASTM D1364 Methyl isobutyl carbinol mg/kg 1000 ASTM D3329 Mesityloxide + mg/kg 1000 ASTM D3329 Isomesityloxide Dimethylketone mg/kg 1000 ASTM D3329 Appearance Cl & FFSM ASTM D4176 Color Pt-Co 10 ASTM D1209 Density @20°C g/mL 0.799 0.802 ASTM D4052 (4) Refractive Index @20°C 1.395 1.397 ASTM D1218 (4) Acidity as Acetic acid % m/m 0.005 ASTM D1613 Non Volatile Matter g/100mL 0.002 ASTM D1353 Distillation, IBP °C 114.0 ASTM D1078 (4) Distillation, DP °C 117.0 ASTM D1078 (4) (1) Guaranteed, (2) Typical, (3) Report Only, (4) Guaranteed spec with typical result Product as produced complies with DIN 53247, ASTM D 1153 and ACS 9th edition. Typical Properties Property Unit Method Value Density @20°C kg/L ASTM D4052 0.800 Cubic Expansion Coefficient @20°C (10^-4)/°C - 12 Refractive Index @20°C - ASTM D1218 1.396 Distillation, IBP °C ASTM D1078 114.0 Distillation, -

Characterization of a Crumb Rubber
Abstract 2415 The National Toxicology Program Synthetic Turf/Recycled Tire Crumb Rubber Research: Characterization of a Crumb Rubber Lot for Use in in Vitro and in Vivo Studies Tim Cristy1, Georgia Roberts2, Brian Burback1, Scott Masten2 and Suramya Waidyanatha2 1Battelle Memorial Institute, Columbus, OH; 2Division of the National Toxicology Program, National Institute of Environmental Health Sciences, Research Triangle Park, NC 27709 ABSTRACT METHODS AND INSTRUMENTATION RESULTS RESULTS ANALYSES OF VOLATILES (PPM) BY HEADSPACE GC-MS Optical Microscopy: An Olympus SZX12 stereo microscope with an Olympus DP-71 camera Artificial turf athletic fields have been growing in popularity due to decreased ANALYSIS FOR INORGANICS a (Tokyo, Japan). Compound Replicate 1 Replicate 2 Average Match Score (%) maintenance required relative to conventional fields. Crumb rubber (CR) used as an Element Replicate 1 Replicate 2 Average Methyl Isobutyl Ketone 3.48 3.21 3.35 98.6 infill in artificial turf has brought public health concerns in recent years. The National Scanning Electron Microscopy (SEM) and Energy Dispersive X-Ray Spectroscopy (EDS): Zinc (%) 1.62 1.74 1.68 Aniline 0.980 1.12 1.05 99.6 JEOL 7600-F (JOEL, Tokyo, Japan) and Apollo X silicon drift detector (EDAX, Mahwah, NJ). Silicon (%) 0.898 0.966 0.932 Benzothiazole 0.753 0.684 0.719 94.0 Toxicology Program (NTP) is conducting research to improve the understanding of Toluene 0.441 0.415 0.428 100.0 Thermogravimetric Analysis: Pyris 1 (Perkin Elmer, Waltham, MA) thermal gravimetric Aluminum (%) -

Chromatographic Separations of Niobium, Tantalum, Molybdenum, and Tungsten Lionel Herbert Dahmer Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1966 Chromatographic separations of niobium, tantalum, molybdenum, and tungsten Lionel Herbert Dahmer Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Analytical Chemistry Commons Recommended Citation Dahmer, Lionel Herbert, "Chromatographic separations of niobium, tantalum, molybdenum, and tungsten " (1966). Retrospective Theses and Dissertations. 5355. https://lib.dr.iastate.edu/rtd/5355 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. This dissertation has been microfihned exactly as received 67-5577 DAHMER, Lionel Herbert, 1936- CHROMATOGRAPHIC SEPARATIONS OF NIOBIUM TANTALUM, MOLYBDENUM, AND TUNGSTEN. Iowa State University of Science and Technology, Ph.D., 1966 Chemistry, analytical University Microfilms, Inc., Ann Arbor, Michigan CHROyiATOGRAPHIC SEPARATIONS OP NIOBIUM TANTALUM, MOLYBDENUM, AND TUNGSTEN by Lionel Herbert Dahmer A Dissertation Submitted to the Graduate Faculty in Partial Fulfillment of The Requirements for the Degree of DOCTOR OF PHILOSOPHY Major Subject: Analytical Chemistry Approved : Signature was redacted for privacy. In Cha lor V/ork Signature was redacted for privacy. Heâi of Major Department Signature was redacted for privacy. De^t of Gr&duate College Iowa State University Of Science and Technology Ames, Iowa 1966 ii TABLE OP CONTEXTS Page INTRODUCTION 1 PART I. CATION EXCHANGE SEPARATION OF MOLYBDENUM, 8 TUNGSTEN, NIOBIUM, AND TANTALUM PROM OTHER METAL IONS LITERATURE SURVEY 9 EXPSRIKSITAL 10 Apparatus 10 Reagents 10 Separation Procedure 12 Analysis of Column Effluents 14 Results and Discussion 15 PART II. -

Silfort* SHC1200 Silfort* SHC1200
Technical Data Sheet SilFORT* SHC1200 SilFORT* SHC1200 Description SilFORT SHC1200 Hard Coat SHC1200 hard coat has been found to yield a clear mar-resistant film when applied to a suitably prepared plastic substrate. It can be applied by flow, dip or spray coating. SilFORT SHP401 Primer SHP401 air-dried primer is used as an adhesion promoter for SHC1200 hard coat on polycarbonate resin. It can be applied by flow, dip or spray coating. Key Features and Benefits Fast cure Abrasion resistance Scratch resistance Good clarity Solvent/chemical resistance SHP401 Primer No thermal cure required Improves coating adhesion Improves water resistance Page 1 of 6 *SilFORT ist ein Markenname der Momentive Performance Materials Inc. SilFORT* SHC1200 Improves ultraviolet resistance SHP401 Primer/SHC1200 Hard Coat on polycarbonate (2 - 4 micron Topcoat Thickness) Cured Film Properties Film Thickness, slow dip coat, 10-18 cm/min 2 – 4 micron withdrawal,18-20% solids at 22C Taber Abrasion, 500 cycles 500G on primed < 6.0 % Haze measured per polycarbonate (CS10F wheel) ASTM D1003. * Index of Refraction 1.4 *Humidity during coating and testing will affect final values. Typical Physical Properties Property SHC1200 Hard Coat SHP401 Primer Solids Content, % 20 ± 1 2.1 ± 0.2 Methanol, Isobutanol, 1-Methoxy-2-propanol, Diacetone Solvent Isopropanol Alcohol Flash Point, PMCC 19.4C 36.1C Density, g/cm3 0.911 0.959 pH 7.3 ± 0.2 - Viscosity, cstk @ 25°C 20 ± 3 4 - 7 Dry Film Thickness, 2.75 - 4.5 0.5 micron VOC, g/l 710 937 Patent Status Nothing contained herein shall be construed to imply the nonexistence of any relevant patents or to constitute the permission, inducement or recommendation to practice any invention covered by any patent, without authority from the owner of the patent. -
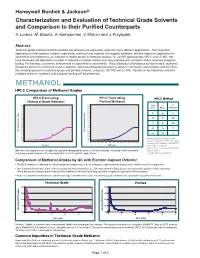
Methanol and Acetone Is in Organic Synthesis
Honeywell Burdick & Jackson® Characterization and Evaluation of Technical Grade Solvents and Comparison to their Purified Counterparts S. Lorenz, M. Bosma, A. Kemperman, V. Mohan and J. Przybytek Abstract: Technical grade solvents and their purified counterparts are separately useful for many different applications. One important application for the common solvents acetonitrile, methanol and acetone is in organic synthesis. Another important application for acetonitrile and methanol is as a diluent or mobile phase in chemical analysis, ie: UV-VIS spectroscopy, HPLC and LC-MS. We have observed and identified a number of impurities in these solvents that may interfere with synthesis and/or accurate analytical testing. For instance, a common contaminant in acetonitrile is acrylonitrile. Trace aldehydes and ketones can be found in methanol. Diacetone alcohol is commonly found in acetone. We have characterized impurity levels in commonly used solvents and will show the variability present in technical grade and purified solvents, using GC, GC-MS and LC-MS. Results on the impurities and their probable effect on synthesis and analytical testing will be presented. METHANOL HPLC Comparison of Methanol Grades HPLC Trace using HPLC Trace using HPLC Method Technical Grade Methanol Purified Methanol 0.200 0.200 Time % % 0.190 0.190 0.180 0.180 (min) Water Methanol 0.170 0.170 0.160 0.160 0.150 0.150 0 95 5 0.140 AU) 0.140 AU) ( ( 0.130 0.130 e e 20 0 100 c 0.120 c 0.120 n 0.110 n 0.110 a a b 0.100 b 0.100 r r 25 0 100 o 0.090 o 0.090 s s 0.080 0.080 -

US EPA, Inert (Other) Pesticide Ingredients in Pesticide Products
Inert Ingredients ordered by CAS Number Updated August 2004 CAS PREFIX NAME List No. 50-21-5 Lactic acid 4B 50-70-4 Sorbitol 4A 50-81-7 L- Ascorbic acid 4A 50-99-7 Dextrose 4A 51-03-6 Piperonyl butoxide 3 51-05-8 Procaine hydrochloride 3 51-55-8 Atropine 3 52-51-7 2- Bromo-2-nitro-propane-1,3-dio 3 54-21-7 Sodium salicylate 3 56-81-5 Glycerol (glycerin) 1,2,3 propanetriol 4A 56-86-0 L- Glutamic acid 3 56-95-1 Chlorhexidine diacetate 3 57-10-3 Hexadecanoic acid 4A 57-11-4 Stearic acid 4A 57-13-6 Urea 4A 57-48-7 D- Fructose 4B 57-50-1 Sugar 4A 57-55-6 Propylene glycol 4B 57-88-5 (3.beta.)- Cholest-5-en-3-ol 4B 58-08-2 1H- Purine-2,6-dione, 3,7-dihydro-1,3,7-trimethyl- 4B 58-56-0 Thiamine mononitrate 4B 58-85-5 Biotin 3 58-86-6 D- Xylose 4B 58-95-7 Vitamin E acetate 3 59-30-3 Folic acid 4B 59-40-5 N-(2- Quinoxalinyl)sulfanilide 3 59-67-6 Nicotinic acid 3 60-00-4 Ethylenediaminetetraacetic acid (EDTA) 4B 60-12-8 Benzeneethanol 3 60-29-7 Ethane, 1,1'-oxybis- 3 60-33-3 Linoleic acid 3 61-73-4 C.I. Basic Blue 9 3 62-33-9 Ethylenediaminetetraacetic acid (EDTA), calcium4B 62-54-4 Acetic acid, calcium salt 4A 63-42-3 D-(+)-Lactose 4A 63-68-3 L- Methionine 4B 64-02-8 Ethylenediaminetetraacetic acid (EDTA), tetraso4B 64-17-5 Ethanol 4B 64-18-6 Formic acid 3 64-19-7 Acetic acid 4B 64-86-8 Colchicine 3 65-85-0 Benzoic acid 4B 66-71-7 1,10- Phenanthroline 3 67-03-8 Thiamin hydrochloride 3 67-43-6 1,1,4,7,7- Diethylenetriaminepentaacetic acid 3 67-48-1 Choline chloride 4B 67-56-1 Methyl alcohol 3 67-63-0 2- Propanol 4B 67-64-1 Acetone 3 67-68-5 Dimethyl -

Safety Assessment of Diacetone Alcohol As Used in Cosmetics
Safety Assessment of Diacetone Alcohol as Used in Cosmetics Status: Draft Tentative Report for Panel Review Release Date: February 16, 2021 Panel Meeting Date: March 11 – 12, 2021 The Expert Panel for Cosmetic Ingredient Safety members are: Chair, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; David E. Cohen, M.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; Lisa A. Peterson, Ph.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. Previous Panel member involved in this assessment: James G. Marks, Jr., M.D. The Cosmetic Ingredient Review (CIR) Executive Director is Bart Heldreth, Ph.D. This safety assessment was prepared by Priya Cherian, Scientific Analyst/Writer, CIR. © Cosmetic Ingredient Review 1620 L Street, NW, Suite 1200 ♢ Washington, DC 20036-4702 ♢ ph 202.331.0651 ♢ fax 202.331.0088 ♢ [email protected] Distributed for Comment Only -- Do Not Cite or Quote Commitment & Credibility since 1976 Memorandum To: Expert Panel for Cosmetic Ingredient Safety Members and Liaisons From: Priya Cherian, Scientific Analyst/Writer, CIR Date: February 16, 2021 Subject: Safety Assessment of Diacetone Alcohol as Used in Cosmetics Enclosed is the Draft Tentative Report of the Safety Assessment of Diacetone Alcohol as Used in Cosmetics (diacet032021rep). At the September 2020 meeting, the Expert Panel for Cosmetic Ingredient Review Safety (Panel) issued an Insufficient Data Announcement (IDA) for this ingredient. In order to come to a conclusion of safety, the Panel requested impurities data. Since the previous review of this report, no new data have been received. -

Trace Analysis of Mesityl Oxide and Diacetone Alcohol in Pharmaceuticals by Capillary Gas Chromatography with Flame Ionization Detection
id22818046 pdfMachine by Broadgun Software - a great PDF writer! - a great PDF creator! - http://www.pdfmachine.com http://www.broadgun.com AASeptemnnbeaar 2ll0y0y9ttiiccaall Volume 8 Issue 3 An Indian Journal Trade Science Inc. CCHHEEMMIISSTTRRYY Full Paper ACAIJ, 8(3) 2009 [346-349] Trace analysis of mesityl oxide and diacetone alcohol in pharmaceuticals by capillary gas chromatography with flame ionization detection K.Rama Seshaiah*1,2, V.Krishna Reddy1, Ch.Krishnaiah1, VVNKV Prasadaraju1, K.Mukkanti2, V.Ranga Reddy1 ’ 1Dr.Reddy s Laboratories Ltd. Active pharmaceutical ingredients, IPDO, Bachupally, Hyderabad-500072, A.P, (INDIA) 2Department of chemisty, J.N.T.University, Kukatpally, Hyderabad-500072, A.P, (INDIA) E-mail: [email protected] Received: 26th May, 2009 ; Accepted: 5th June, 2009 ABSTRACT KEYWORDS A capillary gas chromatographic method using flame ionization detection Pharmaceutical analysis; was developed and validated for the trace analysis (ppm level) of mesityl Mesityl oxide and Diacetone oxide and diacetone alcohol in pharmaceutical drug substance. The method alcohol. utilizes a megabore capillary column with bonded and crosslinked polyeth- ylene glycol stationary phase. A dissolve-and-injection approach was adopted for sample introduction in a split mode (1:1). Water is used as ppm and limit of sample solvent. A limit of detection of about 1.5 and 2 ppm were achieved for the Mesityl oxide and quantitation of about 6 and 10 Diacetone alcohol in drug substance samples. The method optimization and validation are also discussed in this paper. 2009 Trade Science Inc. - INDIA INTRODUCTION ods that are sensitive enough and meet all the regula- tory requirements. Recently, the potential health hazards of trace The pure mesityl oxide and diacetone alcohol are amounts of mesityl oxide and diacetone alcohol in phar- liquids at ambient temperature with a boiling point °C and 170°C respectively.