Dynamics of Transcription Elongation Are Finely Tuned by Dozens of Regulatory Factors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

(P -Value<0.05, Fold Change≥1.4), 4 Vs. 0 Gy Irradiation
Table S1: Significant differentially expressed genes (P -Value<0.05, Fold Change≥1.4), 4 vs. 0 Gy irradiation Genbank Fold Change P -Value Gene Symbol Description Accession Q9F8M7_CARHY (Q9F8M7) DTDP-glucose 4,6-dehydratase (Fragment), partial (9%) 6.70 0.017399678 THC2699065 [THC2719287] 5.53 0.003379195 BC013657 BC013657 Homo sapiens cDNA clone IMAGE:4152983, partial cds. [BC013657] 5.10 0.024641735 THC2750781 Ciliary dynein heavy chain 5 (Axonemal beta dynein heavy chain 5) (HL1). 4.07 0.04353262 DNAH5 [Source:Uniprot/SWISSPROT;Acc:Q8TE73] [ENST00000382416] 3.81 0.002855909 NM_145263 SPATA18 Homo sapiens spermatogenesis associated 18 homolog (rat) (SPATA18), mRNA [NM_145263] AA418814 zw01a02.s1 Soares_NhHMPu_S1 Homo sapiens cDNA clone IMAGE:767978 3', 3.69 0.03203913 AA418814 AA418814 mRNA sequence [AA418814] AL356953 leucine-rich repeat-containing G protein-coupled receptor 6 {Homo sapiens} (exp=0; 3.63 0.0277936 THC2705989 wgp=1; cg=0), partial (4%) [THC2752981] AA484677 ne64a07.s1 NCI_CGAP_Alv1 Homo sapiens cDNA clone IMAGE:909012, mRNA 3.63 0.027098073 AA484677 AA484677 sequence [AA484677] oe06h09.s1 NCI_CGAP_Ov2 Homo sapiens cDNA clone IMAGE:1385153, mRNA sequence 3.48 0.04468495 AA837799 AA837799 [AA837799] Homo sapiens hypothetical protein LOC340109, mRNA (cDNA clone IMAGE:5578073), partial 3.27 0.031178378 BC039509 LOC643401 cds. [BC039509] Homo sapiens Fas (TNF receptor superfamily, member 6) (FAS), transcript variant 1, mRNA 3.24 0.022156298 NM_000043 FAS [NM_000043] 3.20 0.021043295 A_32_P125056 BF803942 CM2-CI0135-021100-477-g08 CI0135 Homo sapiens cDNA, mRNA sequence 3.04 0.043389246 BF803942 BF803942 [BF803942] 3.03 0.002430239 NM_015920 RPS27L Homo sapiens ribosomal protein S27-like (RPS27L), mRNA [NM_015920] Homo sapiens tumor necrosis factor receptor superfamily, member 10c, decoy without an 2.98 0.021202829 NM_003841 TNFRSF10C intracellular domain (TNFRSF10C), mRNA [NM_003841] 2.97 0.03243901 AB002384 C6orf32 Homo sapiens mRNA for KIAA0386 gene, partial cds. -

Atlas Journal
Atlas of Genetics and Cytogenetics in Oncology and Haematology Home Genes Leukemias Tumors Cancer prone Deep Insight Case Reports Portal Journals Teaching X Y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 NA Atlas Journal Atlas Journal versus Atlas Database: the accumulation of the issues of the Journal constitutes the body of the Database/Text-Book. TABLE OF CONTENTS Volume 13, Number 7, July 2009 Previous Issue / Next Issue Genes ABL1 (v-abl Abelson murine leukemia viral oncogene homolog 1) (9q34.1) - updated. Ali G Turhan. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 757-766. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/ABL.html BCL2L12 (BCL2-like 12 (proline-rich)) (19q13.3). Christos Kontos, Hellinida Thomadaki, Andreas Scorilas. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 767-771. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/BCL2L12ID773ch19q13.html BCR (Breakpoint cluster region) (22q11.2) - updated. Ali G Turhan. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 772-779. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/BCR.html ENAH (enabled homolog (Drosophila)) (1q42.12). Paola Nisticò, Francesca Di Modugno. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 780-785. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/ENAHID44148ch1q42.html FGFR2 (fibroblast growth factor receptor 2) (10q26.13). Masaru Katoh. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 786-799. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/FGFR2ID40570ch10q26.html MAPK6 (mitogen-activated protein kinase 6) (15q21.2). Sylvain Meloche. Atlas Genet Cytogenet Oncol Haematol 2009; 13 (7): 800-804. -

Interactome Analyses Revealed That the U1 Snrnp Machinery Overlaps Extensively with the RNAP II Machinery and Contains Multiple
www.nature.com/scientificreports OPEN Interactome analyses revealed that the U1 snRNP machinery overlaps extensively with the RNAP II Received: 12 April 2018 Accepted: 24 May 2018 machinery and contains multiple Published: xx xx xxxx ALS/SMA-causative proteins Binkai Chi1, Jeremy D. O’Connell1,2, Tomohiro Yamazaki1, Jaya Gangopadhyay1, Steven P. Gygi1 & Robin Reed1 Mutations in multiple RNA/DNA binding proteins cause Amyotrophic Lateral Sclerosis (ALS). Included among these are the three members of the FET family (FUS, EWSR1 and TAF15) and the structurally similar MATR3. Here, we characterized the interactomes of these four proteins, revealing that they largely have unique interactors, but share in common an association with U1 snRNP. The latter observation led us to analyze the interactome of the U1 snRNP machinery. Surprisingly, this analysis revealed the interactome contains ~220 components, and of these, >200 are shared with the RNA polymerase II (RNAP II) machinery. Among the shared components are multiple ALS and Spinal muscular Atrophy (SMA)-causative proteins and numerous discrete complexes, including the SMN complex, transcription factor complexes, and RNA processing complexes. Together, our data indicate that the RNAP II/U1 snRNP machinery functions in a wide variety of molecular pathways, and these pathways are candidates for playing roles in ALS/SMA pathogenesis. Te neurodegenerative disease Amyotrophic Lateral Sclerosis (ALS) has no known treatment, and elucidation of disease mechanisms is urgently needed. Tis problem has been especially daunting, as mutations in greater than 30 genes are ALS-causative, and these genes function in numerous cellular pathways1. Tese include mitophagy, autophagy, cytoskeletal dynamics, vesicle transport, DNA damage repair, RNA dysfunction, apoptosis, and pro- tein aggregation2–6. -

Using Long Nanopore Reads to Delineate Structural Variants (Svs)
Using long nanopore reads to delineate structural variants (SVs) in the human genome SVs, including large deletions, duplications, inversions, translocations and copy-number changes are abundant in large genomes, and require long reads for precise characterisation Contact: [email protected] More information at: www.nanoporetech.com and publications.nanoporetech.com Unique Repeat Unique Repeat Unique a) b) a) b) sequence 1 1 sequence 2 2 sequence 3 1,000 60 Short reads Insertions Long A B C D E Reference chromosome 1 40 reads 800 Deletions > 50 bp Short-read assembly 20 Collapsed repeat consensus Unique contig 1 Long-read Bases sequenced (Mb) assembly 600 0 Unique contig 1 Unique contig 3 0 10 20 30 40 V W X Y Z Reference chromosome 2 Single, fully-resolved contig Count Read length (kb) c) > 50 bp 400 chr7 (q33) 7p21.3 15.321.1 15.3 7p14.3 7p14.1 13 11.2 11.21 11.22 11.23 7q21.11 q21.3 7q22.1 7q31.1 7q33 7q34 7q35 36.1 36.3 Scale 50 kb hg38 chr7: 134,550,000 134,600,000 134,650,000 134,700,000 Inversion A D C B E GENCODE v24 comprehensive transcript set (only Basic displayed by default) 200 AKR1B10 AKR1B15 BGPM CALD1 AKR1B15 BGPM Deletion BGPM A B C E AC009276.4 Duplication A B C C C D E 0 1,000 10,000 20,000 30,000 Translocation V W C D E + A B X Y Z Event size (bp) Adapted from Huddleston, J. et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. -

Association Analyses Based on False Discovery Rate Implicate Many New Loci for Coronary Artery Disease
1 Association analyses based on false discovery rate implicate many new loci 2 for coronary artery disease 3 Christopher P. Nelson1,2,61, Anuj Goel3,4,61, Adam S Butterworth5,6,61, Stavroula Kanoni7,8,61, 4 Tom R. Webb1,2, Eirini Marouli7,8, Lingyao Zeng9, Ioanna Ntalla7,8, Florence Y. Lai1,2, Jemma C. 5 Hopewell10, Olga Giannakopoulou7,8, Tao Jiang5, Stephen E. Hamby1,2, Emanuele Di 6 Angelantonio5,6, Themistocles L. Assimes11, Erwin P. Bottinger12, John C. Chambers13,14,15, 7 Robert Clarke10, Colin NA Palmer16,17, Richard M. Cubbon18, Patrick Ellinor19, Raili Ermel20, 8 Evangelos Evangelou21,22, Paul W. Franks23,24,25, Christopher Grace3,4, Dongfeng Gu26, Aroon 9 D. Hingorani27, Joanna M. M. Howson5, Erik Ingelsson28, Adnan Kastrati9, Thorsten Kessler9, 10 Theodosios Kyriakou3,4, Terho Lehtimäki29, Xiangfeng Lu26, Yingchang Lu12,30, Winfried 11 März31,32,33, Ruth McPherson34, Andres Metspalu35, Mar Pujades-Rodriguez36, Arno 12 Ruusalepp20,37, Eric E. Schadt38, Amand F. Schmidt39, Michael J. Sweeting5, Pierre A. 13 Zalloua40,41, Kamal AIGhalayini42, Bernard D. Keavney43,44, Jaspal S. Kooner14,15,45, Ruth J. F. 14 Loos12,46, Riyaz S. Patel47,48, Martin K. Rutter49,50, Maciej Tomaszewski51,52, Ioanna Tzoulaki21,22, 15 Eleftheria Zeggini53, Jeanette Erdmann54,55,56, George Dedoussis57, Johan L. M. 16 Björkegren37,38,58, EPIC-CVD Consortium, CARDIoGRAMplusC4D, The UK Biobank 17 CardioMetabolic Consortium CHD working group, Heribert Schunkert9,61, Martin Farrall3,4,61, 18 John Danesh5,6,59,61, Nilesh J. Samani1,2,61, Hugh Watkins3,4,61, Panos Deloukas7,8,60,61 19 1. Department of Cardiovascular Sciences, University of Leicester, Leicester LE3 9QP, UK 20 2. -

Atlas Journal
Atlas of Genetics and Cytogenetics in Oncology and Haematology Home Genes Leukemias Solid Tumours Cancer-Prone Deep Insight Portal Teaching X Y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 NA Atlas Journal Atlas Journal versus Atlas Database: the accumulation of the issues of the Journal constitutes the body of the Database/Text-Book. TABLE OF CONTENTS Volume 12, Number 6, Nov-Dec 2008 Previous Issue / Next Issue Genes BCL8 (B-cell CLL/lymphoma 8) (15q11). Silvia Rasi, Gianluca Gaidano. Atlas Genet Cytogenet Oncol Haematol 2008; 12 (6): 781-784. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/BCL8ID781ch15q11.html CDC25A (Cell division cycle 25A) (3p21). Dipankar Ray, Hiroaki Kiyokawa. Atlas Genet Cytogenet Oncol Haematol 2008; 12 (6): 785-791. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/CDC25AID40004ch3p21.html CDC73 (cell division cycle 73, Paf1/RNA polymerase II complex component, homolog (S. cerevisiae)) (1q31.2). Leslie Farber, Bin Tean Teh. Atlas Genet Cytogenet Oncol Haematol 2008; 12 (6): 792-797. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/CDC73D181ch1q31.html EIF3C (eukaryotic translation initiation factor 3, subunit C) (16p11.2). Daniel R Scoles. Atlas Genet Cytogenet Oncol Haematol 2008; 12 (6): 798-802. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/EIF3CID44187ch16p11.html ELAC2 (elaC homolog 2 (E. coli)) (17p11.2). Yang Chen, Sean Tavtigian, Donna Shattuck. Atlas Genet Cytogenet Oncol Haematol 2008; 12 (6): 803-806. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/ELAC2ID40437ch17p11.html FOXM1 (forkhead box M1) (12p13). Jamila Laoukili, Monica Alvarez Fernandez, René H Medema. -

12619 CTR9 (D1Z4F) Rabbit Mab
Revision 1 C 0 2 - t CTR9 (D1Z4F) Rabbit mAb a e r o t S Orders: 877-616-CELL (2355) [email protected] 9 Support: 877-678-TECH (8324) 1 6 Web: [email protected] 2 www.cellsignal.com 1 # 3 Trask Lane Danvers Massachusetts 01923 USA For Research Use Only. Not For Use In Diagnostic Procedures. Applications: Reactivity: Sensitivity: MW (kDa): Source/Isotype: UniProt ID: Entrez-Gene Id: WB, IP H M R Endogenous 140 Rabbit IgG Q6PD62 9646 Product Usage Information 7. Farber, L.J. et al. (2010) Mol Carcinog 49, 215-23. Application Dilution Western Blotting 1:1000 Immunoprecipitation 1:50 Storage Supplied in 10 mM sodium HEPES (pH 7.5), 150 mM NaCl, 100 µg/ml BSA, 50% glycerol and less than 0.02% sodium azide. Store at –20°C. Do not aliquot the antibody. Specificity / Sensitivity CTR9 (D1Z4F) Rabbit mAb recognizes endogenous levels of total CTR9 protein. Species Reactivity: Human, Mouse, Rat Species predicted to react based on 100% sequence homology: Hamster, Chicken, Xenopus, Zebrafish, Bovine, Dog, Pig Source / Purification Monoclonal antibody is produced by immunizing animals with a synthetic peptide corresponding to residues near the amino terminus of human CTR9 protein. Background The PAF (RNA polymerase II (RNAPII) associated factor) complex was initially identified in yeast and is comprised of subunits PAF1, Leo1, Ctr9, Cdc73, RTF1 and Ski8 (1,2). The PAF complex plays an important role in transcription initiation and elongation by RNAPII by regulating the establishment of proper histone modifications such as histone H2B ubiquitination and the recruitment of the histone chaperone FACT (facilitates chromatin transcription) (3-5). -
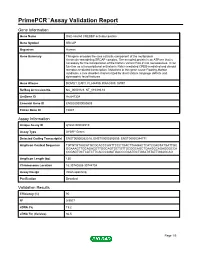
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Snf2-related CREBBP activator protein Gene Symbol SRCAP Organism Human Gene Summary This gene encodes the core catalytic component of the multiprotein chromatin-remodeling SRCAP complex. The encoded protein is an ATPase that is necessary for the incorporation of the histone variant H2A.Z into nucleosomes. It can function as a transcriptional activator in Notch-mediated CREB-mediated and steroid receptor-mediated transcription. Mutations in this gene cause Floating-Harbor syndrome a rare disorder characterized by short stature language deficits and dysmorphic facial features. Gene Aliases DOMO1, EAF1, FLJ44499, KIAA0309, SWR1 RefSeq Accession No. NC_000016.9, NT_010393.16 UniGene ID Hs.647334 Ensembl Gene ID ENSG00000080603 Entrez Gene ID 10847 Assay Information Unique Assay ID qHsaCID0006510 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000262518, ENST00000395059, ENST00000344771 Amplicon Context Sequence TGTGTGTAACATGCGCACCCAGTTCCCTGACTTAAGACTCATCCAGTATGATTGC GGAAAGTTGCAGACGTTGGCAGTGCTGTTGCGGCAGCTCAAGGCAGAGGGCCA CCGAGTGCTCATCTTCACCCAGATGACCCGAATGCTGGATGTATTGGAGCAG Amplicon Length (bp) 130 Chromosome Location 16:30740838-30744704 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 90 R2 0.9977 cDNA Cq 19.2 cDNA Tm (Celsius) 86.5 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report SRCAP, Human -

I STRUCTURAL and FUNCTIONAL CHARACTERIZATION of RTF1
STRUCTURAL AND FUNCTIONAL CHARACTERIZATION OF RTF1 AND INSIGHT INTO ITS ROLE IN TRANSCRIPTIONAL REGULATION by Adam Douglas Wier B.S., Molecular Biology and Biochemistry, Lebanon Valley College, 2009 Submitted to the Graduate Faculty of the Kenneth P. Dietrich School of Arts and Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2014 i UNIVERSITY OF PITTSBURGH KENNETH P. DIETRICH SCHOOL OF ARTS AND SCIENCES This dissertation was presented by Adam D. Wier It was defended on August 20, 2014 and approved by James M. Pipas, Ph.D., Professor, Biological Sciences Karen M. Arndt, Ph.D., Professor, Biological Sciences John M Rosenberg, Ph.D., Professor, Biological Sciences Martin C. Schmidt, Ph.D., Associate Professor, Microbiology and Molecular Genetics Committee Chair: Andrew P. VanDemark, Ph.D., Associate Professor, Biological Sciences ii Copyright © by Adam D. Wier 2014 iii STRUCTURAL AND FUNCTIONAL CHARACTERIZATION OF RTF1 AND INSIGHT INTO ITS ROLE IN TRANSCRIPTIONAL REGULATION Adam D. Wier, PhD University of Pittsburgh, 2014 Originally discovered in a search for RNA polymerase II-associated factors, the Paf1 complex (Paf1C) is best characterized for its roles in regulating transcription elongation. The complex co- localizes with RNA polymerase II from the promoter to the 3’ end of genes and has been linked to a growing list of transcription-related processes including: elongation through chromatin, histone modifications, and recruitment of factors important in transcript maturation. The complex is conserved throughout eukaryotes and is comprised of the proteins Paf1, Ctr9, Cdc73, Rtf1, and Leo1. The domain structures of Paf1C subunits are largely undefined and have few clear homologs, making it difficult to postulate for or localize functions to the individual subunits. -

Unbiased Data Mining Identifies Cell Cycle Transcripts That Predict Non-Indolent Gleason Score 7 Prostate Cancer Wendy L
Johnston et al. BMC Urology (2019) 19:4 https://doi.org/10.1186/s12894-018-0433-5 RESEARCHARTICLE Open Access Unbiased data mining identifies cell cycle transcripts that predict non-indolent Gleason score 7 prostate cancer Wendy L. Johnston1* , Charles N. Catton1,2 and Carol J. Swallow3,4,5,6 Abstract Background: Patients with newly diagnosed non-metastatic prostate adenocarcinoma are typically classified as at low, intermediate, or high risk of disease progression using blood prostate-specific antigen concentration, tumour T category, and tumour pathological Gleason score. Classification is used to both predict clinical outcome and to inform initial management. However, significant heterogeneity is observed in outcome, particularly within the intermediate risk group, and there is an urgent need for additional markers to more accurately hone risk prediction. Recently developed web-based visualization and analysis tools have facilitated rapid interrogation of large transcriptome datasets, and querying broadly across multiple large datasets should identify predictors that are widely applicable. Methods: We used camcAPP, cBioPortal, CRN, and NIH NCI GDC Data Portal to data mine publicly available large prostate cancer datasets. A test set of biomarkers was developed by identifying transcripts that had: 1) altered abundance in prostate cancer, 2) altered expression in patients with Gleason score 7 tumours and biochemical recurrence, 3) correlation of expression with time until biochemical recurrence across three datasets (Cambridge, Stockholm, MSKCC). Transcripts that met these criteria were then examined in a validation dataset (TCGA-PRAD) using univariate and multivariable models to predict biochemical recurrence in patients with Gleason score 7 tumours. Results: Twenty transcripts met the test criteria, and 12 were validated in TCGA-PRAD Gleason score 7 patients.