The Effect of Electrolytes on Kinetics of Oxidation of Sulfite Ion by Ferricyanide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Zinnwald Lithium Project
Zinnwald Lithium Project Report on the Mineral Resource Prepared for Deutsche Lithium GmbH Am St. Niclas Schacht 13 09599 Freiberg Germany Effective date: 2018-09-30 Issue date: 2018-09-30 Zinnwald Lithium Project Report on the Mineral Resource Date and signature page According to NI 43-101 requirements the „Qualified Persons“ for this report are EurGeol. Dr. Wolf-Dietrich Bock and EurGeol. Kersten Kühn. The effective date of this report is 30 September 2018. ……………………………….. Signed on 30 September 2018 EurGeol. Dr. Wolf-Dietrich Bock Consulting Geologist ……………………………….. Signed on 30 September 2018 EurGeol. Kersten Kühn Mining Geologist Date: Page: 2018-09-30 2/219 Zinnwald Lithium Project Report on the Mineral Resource TABLE OF CONTENTS Page Date and signature page .............................................................................................................. 2 1 Summary .......................................................................................................................... 14 1.1 Property Description and Ownership ........................................................................ 14 1.2 Geology and mineralization ...................................................................................... 14 1.3 Exploration status .................................................................................................... 15 1.4 Resource estimates ................................................................................................. 16 1.5 Conclusions and Recommendations ....................................................................... -

US 2004/0237384 A1 Orr (43) Pub
US 2004O237384A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0237384 A1 Orr (43) Pub. Date: Dec. 2, 2004 (54) FUEL COMPOSITIONS EXHIBITING (52) U.S. Cl. ................. 44/314; 44/320; 44/358; 44/359; IMPROVED FUEL STABILITY 44/360; 44/444 (76) Inventor: William C. Orr, Denver, CO (US) Correspondence Address: (57)57 ABSTRACT HOGAN & HARTSON LLP ONE TABOR CENTER, SUITE 1500 A fuel composition of the present invention exhibits mini 1200 SEVENTEENTH ST mized hydrolysis and increased fuel Stability, even after DENVER, CO 80202 (US) extended storage at 65 F. for 6–9 months. The composition, which is preferably not strongly alkaline (3.0 to 10.5), is (21) Appl. No.: 10/722,063 more preferably weakly alkaline to mildly acidic (4.5 to 8.5) (22) Filed: Nov. 24, 2003 and most preferably slightly acidic (6.3 to 6.8), includes a e ars lower dialkyl carbonate, a combustion improving amount of Related U.S. Application Data at least one high heating combustible compound containing at least one element Selected from the group consisting of (63) Continuation-in-part of application No. 08/986,891, aluminum, boron, bromine, bismuth, beryllium, calcium, filed on Dec. 8, 1997, now Pat. No. 6,652,608. cesium, chromium, cobalt, copper, francium, gallium, ger manium, iodine, iron, indium, lithium, magnesium, manga Publication Classification nese, molybdenum, nickel, niobium, nitrogen, phosphorus, potassium, palladium, rubidium, Sodium, tin, Zinc, (51) Int. Cl." ........ C10L 1/12; C1OL 1/30; C1OL 1/28; praseodymium, rhenium, Silicon, Vanadium, or mixture, and C1OL 1/18 a hydrocarbon base fuel. -

Local Peralkalinity in Peraluminous Granitic Pegmatites. I. Evidence
This is the peer-reviewed, final accepted version for American Mineralogist, published by the Mineralogical Society of America. The published version is subject to change. Cite as Authors (Year) Title. American Mineralogist, in press. DOI: https://doi.org/10.2138/am-2021-7790. http://www.minsocam.org/ Revision 1 1 Local peralkalinity in peraluminous granitic pegmatites. I. Evidence 2 from whewellite and hydrogen carbonate in fluid inclusions 3 YONGCHAO LIU1,2, CHRISTIAN SCHMIDT2,*, AND JIANKANG LI1 4 1MNR Key Laboratory of Metallogeny and Mineral Assessment, Institute of Mineral Resources, 5 Chinese Academy of Geological Sciences, Beijing 100037, China 6 2GFZ German Research Centre for Geosciences, Telegrafenberg, 14473 Potsdam, Germany 7 *Corresponding author: [email protected] 8 9 ABSTRACT 10 Fluid inclusions in pegmatite minerals were studied using Raman spectroscopy to 11 determine the carbon species. Carbon dioxide is very abundant in the aqueous liquid and vapor − 12 phases. Occasionally, CH4 was found in the vapor. In the aqueous liquid, HCO3 was detected in 13 fluid inclusions in tantalite-(Mn) from the Morrua Mine and in late-stage quartz from the Muiâne 14 pegmatite and the Naipa Mine, all in the Alto Ligonha District, Mozambique. Moreover, we 15 observed a carbonate (calcite group) in fluid inclusions in garnet from the Naipa Mine and in 16 beryl from the Morrua Mine, both in the Alto Ligonha District, Mozambique, and a calcite-group 17 carbonate and whewellite [CaC2O4∙H2O] in fluid inclusions in topaz from Khoroshiv, Ukraine. 18 The occurrence of oxalate is interpreted to be due to a reaction of some form of carbon (possibly 19 CO or bitumen) with a peralkaline fluid. -

Lithium Resources and Requirements by the Year 2000
Lithium Resources and Requirements by the Year 2000 GEOLOGICAL SURVEY PROFESSIONAL PAPER 1005 Lithium Resources and Requirements by the Year 2000 JAMES D. VINE, Editor GEOLOGICAL SURVEY PROFESSIONAL PAPER 1005 A collection of papers presented at a symposium held in Golden, Colorado, January 22-24, 1976 UNITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1976 UNITED STATES DEPARTMENT OF THE INTERIOR THOMAS S. KLEPPE, Secretary GEOLOGICAL SURVEY V. E. McKelvey, Director First printing 1976 Second printing 1977 Library of Congress Cataloging in Publication Data Vine, James David, 1921- Lithium resources and requirements by the year 2000. (Geological Survey Professional Paper 1005) 1. Lithium ores-United States-Congresses. 2. Lithium-Congresses. I. Vine, James David, 1921- II. Title. HI. Series: United States Geological Survey Professional Paper 1005. TN490.L5L57 553'.499 76-608206 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 Stock Number 024-001-02887-5 CONTENTS Page 1. Introduction, by James D. Vine, U.S. Geological Survey, Denver, Colo ______________-_______-_-- — ------- —— —— ——— ---- 1 2. Battery research sponsored by the U.S. Energy Research and Development Administration, by Albert Landgrebe, Energy Research and De velopment Administration, Washington, D.C., and Paul A. Nelson, Argonne National Laboratory, Argonne, Ill-__- —— -____.—————— 2 3. Battery systems for load-leveling and electric-vehicle application, near-term and advanced technology (abstract), by N. P. Yao and W. J. Walsh, Argonne National Laboratory, Argonne, 111___.__________________________________-___-_________ — ________ 5 4. Lithium requirements for high-energy lithium-aluminum/iron-sulfide batteries for load-leveling and electric-vehicle applications, by A. -

I I W O 2U12/U35556 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date i i 22 March 2012 (22.03.2012) W O 2U12/U35556 Al (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C08G 73/18 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, (21) International Application Number: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, PCT/IN20 11/000628 DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 14 September 201 1 (14.09.201 1) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, (26) Publication Langi English RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, (30) Priority Data: ZM, ZW. 2171/DEL/2010 14 September 2010 (14.09.2010) IN (84) Designated States (unless otherwise indicated, for every (71) Applicant (for all designated States except US): COUN¬ kind of regional protection available): ARIPO (BW, GH, CIL OF SCIENTIFIC & INDUSTRIAL RESEARCH GM, KE, LR, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, [IN/IN]; Anusandhan Bhawan, Rafi Marg, New Delhi 110 ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, MD, RU, TJ, 001 (IN). -

Spodumene: the Lithium Market, Resources and Processes
minerals Review Spodumene: The Lithium Market, Resources and Processes Colin Dessemond 1, Francis Lajoie-Leroux 1, Gervais Soucy 1,* , Nicolas Laroche 2 and Jean-François Magnan 2 1 Département de génie chimique et de génie biotechnologique, Université de Sherbrooke, Sherbrooke, QC J1K 2R1, Canada; [email protected] (C.D.); [email protected] (F.L.-L.) 2 Nemaska Lithium Inc., Quebec, QC G1K 3X2, Canada; [email protected] (N.L.); [email protected] (J.-F.M.) * Correspondence: [email protected]; Tel.: +1-819-821-8000 (ext: 62167) Received: 17 April 2019; Accepted: 24 May 2019; Published: 29 May 2019 Abstract: This literature review gives an overview of the lithium industry, including the lithium market, global resources, and processes of lithium compounds production. It focuses on the production of lithium compounds from spodumene minerals. Spodumene is one of the most critical minerals nowadays, due to its high lithium content and high rate of extraction. Lithium is one of the most sought-after metals, due to the ever-growing demand for lithium-ion batteries (LiBs). The data on lithium extraction from minerals is scattered through years of patents, journal articles, and proceedings; hence, requiring an in-depth review, including the comprehension of the spodumene phase system, the phase conversion processes, and the lithium extraction processes. Keywords: lithium review; spodumene processes; thermodynamic of spodumene; lithium extraction 1. Introduction Lithium is the third element of the periodic table. It is the lightest of all solid elements (d = 0.53 g cm 3 at 20 C), has the highest specific heat capacity, the smallest ionic radius of all · − ◦ the alkali metals, and a high electrochemical potential [1]. -

Geometallurgical Approach to Understand How the Variability In
Geometallurgical approach to understand how the variability in mineralogy at Zinkgruvan orebodies affects the need for copper activation in the bulk rougher- scavenger flotation Ivan Belo Fernandes Geosciences, master's level (120 credits) 2017 Luleå University of Technology Department of Civil, Environmental and Natural Resources Engineering ABSTRACT Zinkgruvan is a Pb-Zn-Ag deposit located in south-central Sweden, owned and operated by Lundin Mining. The ore is beneficiated by a collective-selective flotation circuit, recovering both galena and sphalerite in a bulk rougher-scavenger flotation stage and later on separating them into two final products. Opportunities for increase in zinc recovery in the bulk rougher-scavenger flotation stage have been identified as the plant is relying on natural Pb-activation to process the ore. Process mineralogical tools were used to characterize four different orebodies from Zinkgruvan (Burkland, Borta Bakom, Nygruvan and Sävsjön) and evaluate the metallurgical performance for flotation and magnetic separation, following a geometallurgical approach to better understand and predict the behavior of such ore types in processing plant. The first hypothesis in this thesis is that by addition of copper sulfate and increased collector dosage, Zn recovery will be improved without being detrimental to galena flotation. Results demonstrated that there is a significant increase in Zn recovery by further increasing collector dosage and copper- activating the flotation pulp in the scavenger stage. For instance, an increase in zinc recovery up to 16% has been achieved after addition of copper sulfate. Galena is readily floatable while sphalerite takes longer to be recovered. In addition, iron sulfides take longer to be recovered and, after addition of copper sulfate, there was an increase in iron sulfide recovery. -

HYSYS OLI Interface
HYSYS® 2004.2 OLI Interface Reference Guide Copyright October 2005 Copyright © 1981-2005 by Aspen Technology, Inc. All rights reserved. Aspen Accounting.21™, Aspen ACM Model Export, Aspen ACOL™, Aspen ACX™ Upgrade to ACOL™, Aspen Adsim®, Aspen Advisor™, Aspen Aerotran®, Aspen Alarm & Event™, Aspen APLE™, Aspen Apollo™, Aspen AtOMS™, Aspen Batch and Event Extractor, Aspen Batch Plus®, Aspen Batch.21™, Aspen Batch.21™ CBT, Aspen BatchCAD™, Aspen BatchSep™, Aspen Blend Model Library™, Aspen Blend™, Aspen BP Crude Oil Database, Aspen Calc CBT, Aspen Calc™, Aspen Capable-to-Promise®, Aspen CatRef®, Aspen Chromatography®, Aspen Cim-IO Core™, Aspen Cim-IO™ for @AGlance, Aspen Cim-IO™ for ABB 1180/ 1190 via DIU, Aspen Cim-IO™ for Bailey SemAPI, Aspen Cim-IO™ for DDE, Aspen Cim-IO™ for Eurotherm Gauge via DCP, Aspen Cim-IO™ for Fisher-Rosemount Chip, Aspen Cim-IO™ for Fisher-Rosemount RNI, Aspen Cim-IO™ for Foxboro FOXAPI, Aspen Cim-IO™ for G2, Aspen Cim-IO™ for GE FANUC via HCT, Aspen Cim-IO™ for Hitachi Ex Series, Aspen Cim-IO™ for Honeywell TDC 3000 via HTL/access, Aspen Cim-IO™ for Intellution Fix, Aspen Cim-IO™ for Measurex MCN, Aspen Cim-IO™ for Measurex ODX, Aspen Cim-IO™ for Moore Apacs via Nim (RNI), Aspen Cim-IO™ for OPC, Aspen Cim-IO™ for PI, Aspen Cim- IO™ for RSLinx, Aspen Cim-IO™ for SetCim/InfoPlus-X/InfoPlus.21, Aspen Cim-IO™ for Toshiba Tosdic, Aspen Cim-IO™ for ULMA 3D, Aspen Cim-IO™ for Westinghouse, Aspen Cim-IO™ for WonderWare InTouch, Aspen Cim-IO™ for Yokogawa ACG10S, Aspen Cim-IO™ for Yokogawa EW3, Aspen Collaborative Forecasting™, -

Exam Name___MULTIPLE CHOICE. Choose the One Alternative That Best Completes The
Exam Name___________________________________ MULTIPLE CHOICE. Choose the one alternative that best completes the statement or answers the question. 1) Which of the following is an ionic compound? 1) A) NO2 B) CF4 C) PCl3 D) SeBr2 E) LiCl 2) Which of the following is an ionic compound? 2) A) CH2O B) Cl2O C) PF5 D) Mg3(PO4)2 E) SCl2 3) Which of the following contains BOTH ionic and covalent bonds? 3) A) CaI2 B) CaSO4 C) COS D) SF6 E) None of the above contain both ionic and covalent bonds. 4) Which of the following is a molecular compound? 4) A) RbBr B) N2O4 C) NaNO3 D) SrSO3 E) CuCl2 5) Which of the following is a molecular compound? 5) A) P4O10 B) ZnS C) NaCN D) SrI2 E) LiOH 6) A covalent bond is best described as: 6) A) a bond between a metal and a polyatomic ion. B) a bond between a metal and a nonmetal. C) a bond between two polyatomic ions. D) the transfer of electrons. E) the sharing of electrons between atoms. 7) An ionic bond is best described as: 7) A) the attraction that holds the atoms together in a polyatomic ion. B) the transfer of electrons from one atom to another. C) the attraction between 2 nonmetal atoms. D) the attraction between 2 metal atoms. E) the sharing of electrons. 8) Which of the following is a molecular element? 8) A)S B)Ag C)Kr D)Mg E)Ti 9) Which of the following is a molecular element? 9) A) Xe B) I C) Li D) Mg E) Ar 1 10) Which of the following is an atomic element? 10) A) Mg B) Br C) O D) H E) N 11) What is the empirical formula for C4H10O2? 11) A) C2H4O B) CHO C) CHO2 D) CH2OE)C2H5O 12) What is the empirical formula for Hg2(NO3)2? 12) A) Hg2NO3 B) Hg(NO3)2 C) Hg4(NO3)4 D) HgNO3 E) Hg2(NO3)2 13) Write the formula for the compound formed between potassium and sulfur. -

Page 1 Note: Within Nine Months from the Publication of the Mention
Europäisches Patentamt (19) European Patent Office & Office européen des brevets (11) EP 0 954 558 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C10L 1/14 (2006.01) C10L 1/10 (2006.01) 14.06.2006 Bulletin 2006/24 C10L 1/02 (2006.01) (21) Application number: 97954909.4 (86) International application number: PCT/US1997/022046 (22) Date of filing: 08.12.1997 (87) International publication number: WO 1998/026028 (18.06.1998 Gazette 1998/24) (54) FUEL COMPOSITIONS EXHIBITING IMPROVED FUEL STABILITY VERBESSERTE BRENNSTOFFESTABILITÄT AUFWEISENDE BRENNSTOFFEZUSAMMENSETZUNGEN COMPOSITIONS DE CARBURANT CARACTERISEES PAR UNE STABILITE AMELIOREE DUDIT CARBURANT (84) Designated Contracting States: (56) References cited: DE FR GB IT NL WO-A-95/23836 WO-A-95/33022 WO-A-96/40844 DE-C- 508 917 (30) Priority: 09.12.1996 US 763696 US-A- 4 390 344 US-A- 4 397 655 US-A- 4 929 252 (43) Date of publication of application: 10.11.1999 Bulletin 1999/45 • DATABASE WPI Section Ch, Week 7626 Derwent Publications Ltd., London, GB; Class E11, AN (73) Proprietor: ORR, William C. 76-48473X XP002065173 & JP 51 052 403 A Denver, (ADACHI M) , 10 May 1976 Colorado 80210 (US) Remarks: (72) Inventor: ORR, William C. The file contains technical information submitted after Denver, the application was filed and not included in this Colorado 80210 (US) specification (74) Representative: Weydert, Robert Dennemeyer & Associates Sàrl P.O. Box 1502 1015 Luxembourg (LU) Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. -
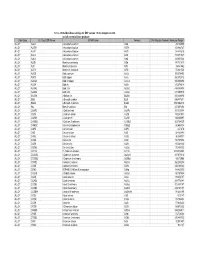
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

Lithium Extraction from Brine with Ion Exchange Resin and Ferric Phosphate
LITHIUM EXTRACTION FROM BRINE WITH ION EXCHANGE RESIN AND FERRIC PHOSPHATE by HIROKI FUKUDA B.Eng., Waseda University, Japan, 2017 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF APPLIED SCIENCE in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Materials Engineering) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) July 2019 © Hiroki Fukuda, 2019 The following individuals certify that they have read, and recommend to the Faculty of Graduate and Postdoctoral Studies for acceptance, a thesis/dissertation entitled: Lithium extraction from brine with ion exchange resin and ferric phosphate submitted by Hiroki Fukuda in partial fulfillment of the requirements for the degree of Master of Applied Science in Materials Engineering Examining Committee: David Dreisinger, Materials Engineering Supervisor David Dixon, Materials Engineering Supervisory Committee Member Berend Wassink, Materials Engineering Supervisory Committee Member Daan Maijer (Chair), Materials Engineering Additional Examiner Additional Supervisory Committee Members: Supervisory Committee Member Supervisory Committee Member - ii - Abstract Lithium is an essential metal for our society. Notably, increasing energy storage system will necessitate much more lithium in the future. This study focused on brine deposit while lithium exists in hard rocks as well. Conventionally, solar evaporation has been used to concentrate lithium from brine, but it takes more than one year. Thus, a more rapid process is desired for the accelerating demand. Here, two types of adsorbent, ion exchange (IX) resin and heterosite ferric phosphate (FP), were studied in order to extract lithium selectively from brine rapidly. First, more than thirty IX resins were tested in lithium chloride solution. Out of the thirty, sulfonate, iminodiacetate and aminomethylphosphonate resins succeeded in extracting lithium with the value of 16.3–32.9 mg-Li/g.