Durham E-Theses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Report of the Advisory Group to Recommend Priorities for the IARC Monographs During 2020–2024
IARC Monographs on the Identification of Carcinogenic Hazards to Humans Report of the Advisory Group to Recommend Priorities for the IARC Monographs during 2020–2024 Report of the Advisory Group to Recommend Priorities for the IARC Monographs during 2020–2024 CONTENTS Introduction ................................................................................................................................... 1 Acetaldehyde (CAS No. 75-07-0) ................................................................................................. 3 Acrolein (CAS No. 107-02-8) ....................................................................................................... 4 Acrylamide (CAS No. 79-06-1) .................................................................................................... 5 Acrylonitrile (CAS No. 107-13-1) ................................................................................................ 6 Aflatoxins (CAS No. 1402-68-2) .................................................................................................. 8 Air pollutants and underlying mechanisms for breast cancer ....................................................... 9 Airborne gram-negative bacterial endotoxins ............................................................................. 10 Alachlor (chloroacetanilide herbicide) (CAS No. 15972-60-8) .................................................. 10 Aluminium (CAS No. 7429-90-5) .............................................................................................. 11 -

Working with Hazardous Chemicals
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

Compound Formula Tin (II) Nitride Silver Oxide Lithium Sulfide Magnesium Sulfide
Ionic Bonding Drill Write the correct formula for the following compounds Compound Formula tin (II) nitride silver oxide lithium sulfide magnesium sulfide copper (I) nitride AgCl boron iodide potassium fluoride copper (I) chloride is CuCl iron (II) oxide is FeO tin (IV) fluoride is SnF4 nickel (II) fluoride is NiF2 lead (IV) oxide is PbO2 silver chloride is calcium iodide is CaI2 potassium bromide sodium phosphide iron (II) chloride copper (I) bromide lead (II) sulfide lead (IV) nitride beryllium nitride potassium bromide is KBr sodium phosphide is Na3P iron (II) chloride is FeCl2 copper (I) bromide is CuBr lead (II) sulfide is PbS lead (IV) nitride is Pb3N4 beryllium nitride is Be3N2 copper (I) chloride iron (II) oxide tin (IV) fluoride nickel (II) fluoride lead (IV) oxide Ag2O silver chloride calcium iodide Answers copper (I) nitride is Cu3N boron iodide is BI3 potassium fluoride is KF silver oxide is lithium sulfide is Li2S magnesium sulfide is MgS tin (II) nitride is Sn3N2 Ionic Bonding Drill Write the correct formula for the following compounds Compound Formula lithium bromide sodium sulfide lead (II) chloride nickel (II) oxide AlBr3 copper (II) oxide AlI3 iron (II) fluoride tin (II) oxide iron (II) oxide is FeO lead (II) oxide is PbO aluminum bromide is potassium oxide is K2O potassium oxide is K2O aluminum iodide is lead (II) nitride is Pb3N2 tin (IV) sulfide iron (III) sulfide lead (II) nitride copper (II) oxide silver fluoride AgF sodium chloride magnesium bromide tin (IV) sulfide is SnS2 iron (III) sulfide is Fe2S3 lead (II) -

Preparing the Future Post-Mortem Analysis of Beryllium-Based JET and ITER Samples by Multi-Wavelengths Raman Spectroscopy on Implanted Be, and Co-Deposited Be M
Preparing the future post-mortem analysis of beryllium-based JET and ITER samples by multi-wavelengths Raman spectroscopy on implanted Be, and co-deposited Be M. Rusu, C. Pardanaud, Y. Ferro, G. Giacometti, C. Martin, Y. Addab, P. Roubin, M. Minissale, L. Ferri, F. Virot, et al. To cite this version: M. Rusu, C. Pardanaud, Y. Ferro, G. Giacometti, C. Martin, et al.. Preparing the future post-mortem analysis of beryllium-based JET and ITER samples by multi-wavelengths Raman spectroscopy on im- planted Be, and co-deposited Be. Nuclear Fusion, IOP Publishing, 2017, 57, pp.076035. 10.1088/1741- 4326/aa70bb. hal-01787067 HAL Id: hal-01787067 https://hal.archives-ouvertes.fr/hal-01787067 Submitted on 7 May 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Preparing the future post-mortem analysis of beryllium-based JET and ITER samples by multi-wavelengths Raman spectroscopy on implanted Be, and co-deposited Be M. I. Rusua,b, C. Pardanaudb*, Y. Ferrob, G. Giacomettib, C. Martinb, Y. Addabb, P. Roubinb, M. Minissaleb,f, L. Ferryb,c, F. Virotc, M. Barrachin c, C P Lungud, C Porosnicud, P Dincad, M Lungud, M. -
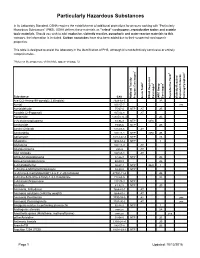
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/074683 Al 19 May 2016 (19.05.2016) W P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C12N 15/10 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/DK20 15/050343 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 11 November 2015 ( 11. 1 1.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: PA 2014 00655 11 November 2014 ( 11. 1 1.2014) DK (84) Designated States (unless otherwise indicated, for every 62/077,933 11 November 2014 ( 11. 11.2014) US kind of regional protection available): ARIPO (BW, GH, 62/202,3 18 7 August 2015 (07.08.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: LUNDORF PEDERSEN MATERIALS APS TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, [DK/DK]; Nordvej 16 B, Himmelev, DK-4000 Roskilde DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, (DK). -

Itetall IC BROMIDES AS CATALYSTS in THE
QUANTITATIVE COMPARISON OF ;itETALL IC BROMIDES AS CATALYSTS IN THE FRIED.liL-CRAFTS KETONE SYNTHESIS i QUANTITATIVE COMPARISON OF MET ALLIC BROMIDES AS CATAL YSTS IN THE FRIEDEL-CRAFTS KETONE SYNTHESIS By Peter Taketoshi M,, ori Bachelor of .Arts Park College Parkville, Missouri 1945 Submitted to the Department of Chemistry Oklahoma Agricultural and Mechanical College In partial fulfillment of the r equirement for t he Degree of Master of Science 194'~ ii \ APPROVED BY: Chairman, Thesis Committee Head of the Department ~he~ ( ~uate~~ School 2174~10 iii ACKNOWLEDGEMENT The author wis hes to express his s i neere gr atitude to Dr . O. C. Dermer under whose guidance t his work has been accomplished . He also wi s hes to express hi s a ppreciation to the Chemi stry Department for t he s uppl y of chemicals used. iv TABLE OF CONTENTS "P age Introduction • • l Historical • • • • 2 Experimental • • • • 5 Procadure • • • • • • • • 9 Table of results . • • • • • • • 12 Discussion of results • .. • • • • • • 15 Summary • • • • • • • • 22 Bibliography • • • • • • • • • • 23 Biography • • • • • • • • • • • 25 1 I NTRODCCT ION T~lis is a continuation of the s t udy of catalystf for the Friedel Crafts ketone synt hesis star ted by Wilson ( .38 ), a nd continued by Suguj. tan (34), Johnson (17), and Billme ier (4). Ma ny metallic chlorides have been used in t his reacti on, but metallic bromides, as cat alyst s, have been rather neelected . It is the purpose of this work to study the ef fectiveness of some of the metallic bromides by following essentially the experimental pro cedure of Billmeier (4). 2 HISTORICAL In 1877, the French chemist Friedel and his American colleague Crafts (12) discovered the f amous Friedel-Crafts reaction, which now has s uch great industrial a pplication (7 , 13, 18, 35). -

Complexes of Unsaturated and Other Organoberyllium Compounds
Durham E-Theses Complexes of unsaturated and other organoberyllium compounds Francis, B. R. How to cite: Francis, B. R. (1970) Complexes of unsaturated and other organoberyllium compounds, Durham theses, Durham University. Available at Durham E-Theses Online: http://etheses.dur.ac.uk/8700/ Use policy The full-text may be used and/or reproduced, and given to third parties in any format or medium, without prior permission or charge, for personal research or study, educational, or not-for-prot purposes provided that: • a full bibliographic reference is made to the original source • a link is made to the metadata record in Durham E-Theses • the full-text is not changed in any way The full-text must not be sold in any format or medium without the formal permission of the copyright holders. Please consult the full Durham E-Theses policy for further details. Academic Support Oce, Durham University, University Oce, Old Elvet, Durham DH1 3HP e-mail: [email protected] Tel: +44 0191 334 6107 http://etheses.dur.ac.uk COMPIEXES OF UNSATURATED AND OTHER ORGANOBERYLLIUM COMPOUNDS BY B. R. FRANCIS, B.Sc. 1— A thesis submitted for the Degree of Doctor of Philosophy in the University of Durham. JANUARY 1970 Ac knowledgeme nts The author wishes to express his sincere thanks to Professor G.E.Coates, NI.A., D.Sc, F.R.I.C., under whose supervision this research was carried out, for his advice and encouragement both personally and academically, and also to many members of the Chemistry Departments of the University of Durham and the University of Wyoming for their help, enthusiasm and friendship. -

Attachment 3-1 Guidance for Developing Ecological Soil
Attachment 3-1 Guidance for Developing Ecological Soil Screening Levels (Eco-SSLs) Eco-SSL Standard Operating Procedure (SOP # 1): Plant and Soil Invertebrate Literature Search and Acquisition OSWER Directive 92857-55 November 2003 This page intentionally left blank OVERVIEW Currently, there is a lack of clear guidance in setting terrestrial effect thresholds when conducting risk assessments. Without an EPA-approved, peer-reviewed, ecologically-based terrestrial effect database, the process to develop thresholds is problematic both to EPA, other federal agencies, states, and concerned private parties. Identification of published toxicity studies on invertebrates, microbial processes and plants is a key step in the derivation of benchmarks. The purpose of the Task Group 4, Standard Operating Procedure Number 1: Literature Search and Acquisition (referred to as TG4-SOP#1) is to document procedures used to identify and acquire potentially relevant toxicology literature for use in setting ecological soil screening levels. The literature search strategy is designed to locate worldwide terrestrial toxicity literature that includes the effects of chemicals of concern on terrestrial soil-dwelling invertebrates and plants. The literature acquisition process is designed to ensure timely acquisition of relevant publications. LITERATURE IDENTIFICATION Potentially relevant literature for developing ecological soil screening levels (Eco-SSLs) is identified by examining hard copies of relevant journals, bibliographies and guidance publications and through the use of a comprehensive computerized literature search strategy. These procedures are designed to locate worldwide terrestrial toxicology literature that includes the effects of specific toxic substances with an emphasis on exposure via soil. Paper-based Literature Identification The paper-based literature identification process includes the scanning of relevant review article bibliographies and key journals held in the U.S. -

Technical Background Document (U.S
Part 5: CHEMICAL-SPECIFIC PARAMETERS Chemical-specific parameters required for calculating soil screening levels include the organic carbon normalized soil-water partition coefficient for organic compounds (Koc), the soil-water partition coefficient for inorganic constituents (Kd), water solubility (S), Henry's law constant (HLC, HN), air diffusivity (Di,a), and water diffusivity (Di,w). In addition, the octanol-water partition coefficient (Kow) is needed to calculate Koc values. This part of the background document describes the collection and compilation of these parameters for the SSL chemicals. With the exception of values for air diffusivity (Di,a), water diffusivity (Di,w), and certain Koc values, all of the values used in the development of SSLs can be found in the Superfund Chemical Data Matrix (SCDM). SCDM is a computer code that includes more than 25 datafiles containing specific chemical parameters used to calculate factor and benchmark values for the Hazard Ranking System (HRS). Because SCDM datafiles are regularly updated, the user should consult the most recent version of SCDM to ensure that the values are up to date. 5.1 Solubility, Henry's Law Constant, and Kow Chemical-specific values for solubility, Henry's law constant (HLC), and Kow were obtained from SCDM. In the selection of the value for SCDM, measured or analytical values are favored over calculated values. However, in the event that a measured value is not available, calculated values are used. Table 36 presents the solubility, Henry's law constant, and Kow values taken from SCDM and used to calculate SSLs. Henry's law constant values were available for all but two of the constituents of interest. -

Export Control Handbook for Chemicals
Export Control Handbook for Chemicals -Dual-use control list -Common Military List -Explosives precursors -Syria restrictive list -Psychotropics and narcotics precursors ARNES-NOVAU, X 2019 EUR 29879 This publication is a Technical report by the Joint Research Centre (JRC), the European Commission’s science and knowledge service. It aims to provide evidence-based scientific support to the European policymaking process. The scientific output expressed does not imply a policy position of the European Commission. Neither the European Commission nor any person acting on behalf of the Commission is responsible for the use that might be made of this publication. Contact information Xavier Arnés-Novau Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-785421 Filippo Sevini Joint Research Centre, Via Enrico Fermi 2749, 21027 Ispra (VA), Italy [email protected] Tel.: +39 0332-786793 EU Science Hub https://ec.europa.eu/jrc JRC 117839 EUR 29879 Print ISBN 978-92-76-11971-5 ISSN 1018-5593 doi:10.2760/844026 PDF ISBN 978-92-76-11970-8 ISSN 1831-9424 doi:10.2760/339232 Luxembourg: Publications Office of the European Union, 2019 © European Atomic Energy Community, 2019 The reuse policy of the European Commission is implemented by Commission Decision 2011/833/EU of 12 December 2011 on the reuse of Commission documents (OJ L 330, 14.12.2011, p. 39). Reuse is authorised, provided the source of the document is acknowledged and its original meaning or message is not distorted. The European Commission shall not be liable for any consequence stemming from the reuse. -

Maine Remedial Action Guidelines (Rags) for Contaminated Sites
Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites (RAGs) Effective Date: May 1, 2021 Approved by: ___________________________ Date: April 27, 2021 David Burns, Director Bureau of Remediation & Waste Management Executive Summary MAINE DEPARTMENT OF ENVIRONMENTAL PROTECTION 17 State House Station | Augusta, Maine 04333-0017 www.maine.gov/dep Maine Department of Environmental Protection Remedial Action Guidelines for Contaminated Sites Contents 1 Disclaimer ...................................................................................................................... 1 2 Introduction and Purpose ............................................................................................... 1 2.1 Purpose ......................................................................................................................................... 1 2.2 Consistency with Superfund Risk Assessment .............................................................................. 1 2.3 When to Use RAGs and When to Develop a Site-Specific Risk Assessment ................................. 1 3 Applicability ................................................................................................................... 2 3.1 Applicable Programs & DEP Approval Process ............................................................................. 2 3.1.1 Uncontrolled Hazardous Substance Sites ............................................................................. 2 3.1.2 Voluntary Response Action Program