1-1 Amino Acids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Side-Chain Liquid Crystal Polymers (SCLCP): Methods and Materials. an Overview
Materials 2009, 2, 95-128; doi:10.3390/ma2010095 OPEN ACCESS materials ISSN 1996-1944 www.mdpi.com/journal/materials Review Side-chain Liquid Crystal Polymers (SCLCP): Methods and Materials. An Overview Tomasz Ganicz * and Włodzimierz Stańczyk Centre of Molecular and Macromolecular Studies, Polish Academy of Science, Sienkiewicza 112, 90- 363 Łódź, Poland; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel. +48-42-684-71-13; Fax: +48-42-684-71-26 Received: 5 February 2009; in revised form: 3 March 2009 / Accepted: 9 March 2009 / Published: 11 March 2009 Abstract: This review focuses on recent developments in the chemistry of side chain liquid crystal polymers. It concentrates on current trends in synthetic methods and novel, well defined structures, supramolecular arrangements, properties, and applications. The review covers literature published in this century, apart from some areas, such as dendritic and elastomeric systems, which have been recently reviewed. Keywords: Liquid crystals, side chain polymers, polymer modification, polymer synthesis, self-assembly. 1. Introduction Over thirty years ago the work of Finkelmann and Ringsdorf [1,2] gave important momentum to synthesis of side chain liquid crystal polymers (SCLCPs), materials which combine the anisotropy of liquid crystalline mesogens with the mechanical properties of polymers. Although there were some earlier attempts [2], their approach of decoupling the motions of a polymer main chain from a mesogen, thus allowed side chain moieties to build up long range ordering (Figure 1). They were able to synthesize polymers with nematic, smectic and cholesteric phases via free radical polymerization of methacryloyl type monomers [3]. -

Heterogeneous Impacts of Protein-Stabilizing Osmolytes On
bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Heterogeneous Impacts of Protein-Stabilizing Osmolytes on Hydrophobic Interaction Mrinmoy Mukherjee and Jagannath Mondal⇤ Tata Institute of Fundamental Research Hyderabad, 500107 India E-mail: [email protected],+914020203091 Abstract Osmolytes’ mechanism of protecting proteins against denaturation is a longstanding puzzle, further complicated by the complex diversities inherent in protein sequences. An emergent approach in understanding osmolytes’ mechanism of action towards biopoly- mer has been to investigate osmolytes’ interplay with hydrophobic interaction, the ma- jor driving force of protein folding. However, the crucial question is whether all these protein-stabilizing osmolytes display a single unified mechanism towards hydrophobic interactions. By simulating the hydrophobic collapse of a macromolecule in aqueous solutions of two such osmoprotectants, Glycine and Trimethyl N-oxide (TMAO), both of which are known to stabilize protein’s folded conformation, we here demonstrate that these two osmolytes can impart mutually contrasting effects towards hydropho- bic interaction. While TMAO preserves its protectant nature across diverse range of polymer-osmolyte interactions, glycine is found to display an interesting cross-over from being a protectant at weaker polymer-osmolyte interaction to a denaturant of hydrophobicity at stronger polymer-osmolyte interactions. A preferential-interaction analysis reveals that a subtle balance of conformation-dependent exclusion/binding of ⇤To whom correspondence should be addressed 1 bioRxiv preprint doi: https://doi.org/10.1101/328922; this version posted May 23, 2018. -

The Role of Side-Chain Branch Position on Thermal Property of Poly-3-Alkylthiophenes
Polymer Chemistry The Role of Side-chain Branch Position on Thermal Property of Poly-3-alkylthiophenes Journal: Polymer Chemistry Manuscript ID PY-ART-07-2019-001026.R2 Article Type: Paper Date Submitted by the 02-Oct-2019 Author: Complete List of Authors: Gu, Xiaodan; University of Southern Mississippi, School of Polymer Science and Engineering Cao, Zhiqiang; University of Southern Mississippi, School of Polymer Science and Engineering Galuska, Luke; University of Southern Mississippi, School of Polymer Science and Engineering Qian, Zhiyuan; University of Southern Mississippi, School of Polymer Science and Engineering Zhang, Song; University of Southern Mississippi, School of Polymer Science and Engineering Huang, Lifeng; University of Southern Mississippi, School of Polymers and High Performance Materials Prine, Nathaniel; University of Southern Mississippi, School of Polymer Science and Engineering Li, Tianyu; Oak Ridge National Laboratory; University of Tennessee Knoxville He, Youjun; Oak Ridge National Laboratory Hong, Kunlun; Oak Ridge National Laboratory, Center for Nanophase Materials Science; Oak Ridge National Laboratory, Center for Nanophase Materials Science Page 1 of 13 PleasePolymer do not Chemistryadjust margins ARTICLE The Role of Side-chain Branch Position on Thermal Property of Poly-3-alkylthiophenes Received 00th January 20xx, a a a a a a Accepted 00th January 20xx Zhiqiang Cao, Luke Galuska, Zhiyuan Qian, Song Zhang, Lifeng Huang, Nathaniel Prine, Tianyu Li,b, c Youjun He,b Kunlun Hong,*b, c and Xiaodan Gu*a DOI: 10.1039/x0xx00000x Thermomechanical properties of conjugated polymers (CPs) are greatly influenced by both their microstructures and backbone structures. In the present work, to investigate the effect of side-chain branch position on the backbone’s mobility and molecular packing structure, four poly (3-alkylthiophene-2, 5-diyl) derivatives (P3ATs) with different side chains, either branched and linear, were synthesized by a quasi-living Kumada catalyst transfer polymerization (KCTP) method. -

Protein Structure & Folding
6 Protein Structure & Folding To understand protein folding In the last chapter we learned that proteins are composed of amino acids Goal as a chemical equilibrium. linked together by peptide bonds. We also learned that the twenty amino acids display a wide range of chemical properties. In this chapter we will see Objectives that how a protein folds is determined by its amino acid sequence and that the three-dimensional shape of a folded protein determines its function by After this chapter, you should be able to: the way it positions these amino acids. Finally, we will see that proteins fold • describe the four levels of protein because doing so minimizes Gibbs free energy and that this minimization structure and the thermodynamic involves both making the most favorable bonds and maximizing disorder. forces that stabilize them. • explain how entropy (S) and enthalpy Proteins exhibit four levels of structure (H) contribute to Gibbs free energy. • use the equation ΔG = ΔH – TΔS The structure of proteins can be broken down into four levels of to determine the dependence of organization. The first is primary structure, the linear sequence of amino the favorability of a reaction on acids in the polypeptide chain. By convention, the primary sequence is temperature. written in order from the amino acid at the N-terminus (by convention • explain the hydrophobic effect and its usually on the left) to the amino acid at the C-terminus. The second level role in protein folding. of protein structure, secondary structure, is the local conformation adopted by stretches of contiguous amino acids. -
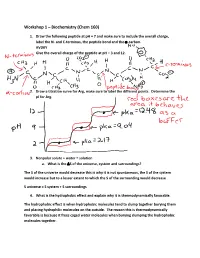
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

Improved Prediction of Protein Side-Chain Conformations with SCWRL4 Georgii G
proteins STRUCTURE O FUNCTION O BIOINFORMATICS Improved prediction of protein side-chain conformations with SCWRL4 Georgii G. Krivov,1,2 Maxim V. Shapovalov,1 and Roland L. Dunbrack Jr.1* 1 Institute for Cancer Research, Fox Chase Cancer Center, 333 Cottman Avenue, Philadelphia, Pennsylvania 19111 2 Department of Applied Mathematics, Moscow Engineering Physics Institute (MEPhI), Kashirskoe Shosse 31, Moscow 115409, Russian Federation INTRODUCTION ABSTRACT The side-chain conformation prediction problem is an integral Determination of side-chain conformations is an component of protein structure determination, protein structure important step in protein structure prediction and protein design. Many such methods have prediction, and protein design. In single-site mutants and in closely been presented, although only a small number related proteins, the backbone often changes little and structure pre- 1 are in widespread use. SCWRL is one such diction can be accomplished by accurate side-chain prediction. In method, and the SCWRL3 program (2003) has docking of ligands and other proteins, taking into account changes remained popular because of its speed, accuracy, in side-chain conformation is often critical to accurate structure pre- and ease-of-use for the purpose of homology dictions of complexes.2–4 Even in methods that take account of modeling. However, higher accuracy at compara- changes in backbone conformation, one step in the process is recal- ble speed is desirable. This has been achieved in culation of side-chain conformation or ‘‘repacking.’’5 Because many a new program SCWRL4 through: (1) a new backbone conformations may be sampled in model refinements, backbone-dependent rotamer library based on side-chain prediction must also be very fast. -

Molecular Modelling Virtual Chemistry Resource Pymol Experiment Answer Sheet Well Done for Completing Our Pymol Experiment! We Really Hope You Enjoyed It!
#Outtheboxthinking Faculty of Natural & Mathematical Sciences Department of Chemistry Molecular Modelling Virtual Chemistry Resource PyMol Experiment Answer Sheet Well done for completing our PyMol experiment! We really hope you enjoyed it! Please make sure you have completed our post experiment questionnaire. https://kings.onlinesurveys.ac.uk/post-questionnaire-for-outtheboxthinking-resources Send us photos, comments, or thoughts to either our twitter or Instagram accounts using the hashtag #outtheboxthinking. We also encourage you to make a poster of your data and email it to us. Exploring how proteins are formed Press “Amino Acid” Q. What is the R group in the side chain of this amino acid? CH3- Methyl Group Q. Which amino acid is shown here? Alanine Press the button “ALA_ASP” and an additional amino acid will appear Q. What is the chemical formula of the side chain of the new amino acid? CH2COOH Q. What is the name of the second, new amino acid? Aspartic Acid or Aspartate Press the button “Bonded_Ala_Asp” Q. What colour(s) is the amide bond connecting the two amino acids? Blue and green. It is important to highlight the bond between the nitrogen of one amino acid and the carbonyl of the second amino acid and not any other bond (Figure 1). Figure 1: Arrow points to the amide bond formed between two amino acids. Page 1 of 8 Instagram: kclchemoutreach #outtheboxthinking Q. Which elements have been lost from each amino acid following their bonding? Amino acid 1:: Alanine has lost an oxygen and a hydrogen Amino acid 2: Aspartic Acid has lost a hydrogen If you are unsure why this is correct look at figure 2 and open the PyMol document again to visualise the bonding Figure 2: Reaction scheme for amino acid bonding, resulting in the formation of an amide bond and the loss of water. -

Amino Acid Recognition by Aminoacyl-Trna Synthetases
www.nature.com/scientificreports OPEN The structural basis of the genetic code: amino acid recognition by aminoacyl‑tRNA synthetases Florian Kaiser1,2,4*, Sarah Krautwurst3,4, Sebastian Salentin1, V. Joachim Haupt1,2, Christoph Leberecht3, Sebastian Bittrich3, Dirk Labudde3 & Michael Schroeder1 Storage and directed transfer of information is the key requirement for the development of life. Yet any information stored on our genes is useless without its correct interpretation. The genetic code defnes the rule set to decode this information. Aminoacyl-tRNA synthetases are at the heart of this process. We extensively characterize how these enzymes distinguish all natural amino acids based on the computational analysis of crystallographic structure data. The results of this meta-analysis show that the correct read-out of genetic information is a delicate interplay between the composition of the binding site, non-covalent interactions, error correction mechanisms, and steric efects. One of the most profound open questions in biology is how the genetic code was established. While proteins are encoded by nucleic acid blueprints, decoding this information in turn requires proteins. Te emergence of this self-referencing system poses a chicken-or-egg dilemma and its origin is still heavily debated 1,2. Aminoacyl-tRNA synthetases (aaRSs) implement the correct assignment of amino acids to their codons and are thus inherently connected to the emergence of genetic coding. Tese enzymes link tRNA molecules with their amino acid cargo and are consequently vital for protein biosynthesis. Beside the correct recognition of tRNA features3, highly specifc non-covalent interactions in the binding sites of aaRSs are required to correctly detect the designated amino acid4–7 and to prevent errors in biosynthesis5,8. -

2. Proteins Have Hierarchies of Structure
27 2. Proteins have Hierarchies of Structure Protein structure is usually described at four different levels (Fig. II.2.1). The first level, called the primary structure, describes the linear sequence of the amino acids in the chain. The different primary structures correspond to the different sequences in which the amino acids are covalently linked together. The secondary structure describes two common patterns of structural repetition in proteins: the coiling up into helices of segments of the chain, and the pairing together of strands of the chain into β-sheets. The tertiary structure is the next higher level of organization, the overall arrangement of secondary structural elements. The quaternary structure describes how different polypeptide chains are assembled into complexes. Figure II.2.1. Different levels of protein structure. A protein chain’s primary structure is its amino acid sequence. Secondary structure consists of the regular organization of helices and sheets. The example shown is a schematic representation of an α helix. Tertiary structure is a polypeptide chain’s three- dimensional native conformation, which often involves compact packing of secondary structure elements. The example given in the figure is a schematic drawing of one of the four polypeptide chains (subunits) of hemoglobin, the protein that transports oxygen in the blood. α-helices along the chain are represented as cylinders. N is the amino terminus and C is the carboxyl terminus of the polypeptide chain. Quaternary structure is the arrangement of multiple polypeptide chains (subunits) to form a functional biomolecular structure. The figure shows the quaternary arrangement of four subunits to form the functional hemoglobin molecule. -

Hydrophobic Effect, Water Structure, and Heat Capacity Changes
J. Phys. Chem. B 1997, 101, 4343-4348 4343 Hydrophobic Effect, Water Structure, and Heat Capacity Changes Kim A. Sharp* and Bhupinder Madan Department of Biochemistry & Biophysics, UniVersity of PennsylVania, 3700 Hamilton Walk, Philadelphia, PennsylVania 19104-6059 ReceiVed: January 16, 1997; In Final Form: April 1, 1997X hyd The hydration heat capacity (∆Cp ) of nine solutes of varying hydrophobicity was studied using a combination of a random network model equation of water and Monte Carlo simulations. Nonpolar solutes cause a concerted decrease in the average length and angle of the water-water hydrogen bonds in the first hydration shell, while polar and ionic solutes have the opposite effect. This is due to changes in the amounts, relative to bulk water, of two populations of hydrogen bonds: one with shorter and more linear bonds and the other with longer and more bent bonds. Heat capacity changes were calculated from these changes in water structure using a random network model equation of state. The calculated changes account for observed hyd differences in ∆Cp for the various solutes. The simulations provide a unified picture of hydrophobic and hyd polar hydration, and both a structural and thermodynamic explanation of the opposite signs of ∆Cp observed for polar and nonpolar solutes. Introduction nonpolar groups means that at higher temperatures (T > 80 °C) their insolubility results from unfavorable changes in enthalpy, In recent years analysis of heat capacity changes has become not entropy. Second, the hydration of a majority of polar and of central importance in understanding the thermodynamics of ionic solutes is also accompanied by a decrease in entropy, and protein folding, protein-protein binding, protein-nucleic acid thus by some kind of structuring of water, but in this case binding, and the hydrophobic effect.1-7 There are several hyd 7,16-20 reasons for this. -

Biological Membranes
14 Biological Membranes To understand the structure The fundamental unit of life is the cell. All living things are composed of Goal and composition of biological cells, be it a single cell in the case of many microorganisms or a highly membranes. organized ensemble of myriad cell types in the case of multicellular organisms. A defining feature of the cell is a membrane, the cytoplasmic Objectives membrane, that surrounds the cell and separates the inside of the cell, the After this chapter, you should be able to cytoplasm, from other cells and the extracellular milieu. Membranes also • distinguish between cis and trans surround specialized compartments inside of cells known as organelles. unsaturated fatty acids. Whereas cells are typically several microns (μm) in diameter (although • explain why phospholipids some cells can be much larger), the membrane is only about 10 nanometers spontaneously form lipid bilayers and (nm) thick. Yet, and as we will see in subsequent chapters, the membrane is sealed compartments. not simply an ultra-thin, pliable sheet that encases the cytoplasm. Rather, • describe membrane fluidity and how it membranes are dynamic structures that mediate many functions in the is affected by membrane composition life of the cell. In this chapter we examine the composition of membranes, and temperature. their assembly, the forces that stabilize them, and the chemical and physical • explain the role of cholesterol in properties that influence their function. buffering membrane fluidity. The preceding chapters have focused on two kinds of biological molecules, • explain how the polar backbone namely proteins and nucleic acids, that are important in the workings of a membrane protein can be accommodated in a bilayer. -

Hydrophobic Catalysis and a Potential Biological Role of DNA Unstacking Induced by Environment Effects
Hydrophobic catalysis and a potential biological role of DNA unstacking induced by environment effects Bobo Fenga,1, Robert P. Sosab,c,d, Anna K. F. Mårtenssona, Kai Jiange, Alex Tongf, Kevin D. Dorfmang, Masayuki Takahashih, Per Lincolna, Carlos J. Bustamanteb,c,d,f,i,j,k,l, Fredrik Westerlunde, and Bengt Nordéna,1 aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden; bBiophysics Graduate Group, University of California, Berkeley, CA 94720; cJason L. Choy Laboratory of Single Molecule Biophysics, University of California, Berkeley, CA 94720; dInstitute for Quantitative Biosciences-QB3, University of California, Berkeley, CA 94720; eDepartment of Biology and Biological Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden; fDepartment of Chemistry, University of California, Berkeley, CA 94720; gDepartment of Chemical Engineering and Materials Science, University of Minnesota, Twin Cities, Minneapolis, MN 55455; hSchool of Life Science and Technology, Tokyo Institute of Technology, Tokyo 152-8550, Japan; iDepartment of Molecular & Cell Biology, University of California, Berkeley, CA 94720; jDepartment of Physics, University of California, Berkeley, CA 94720; kHoward Hughes Medical Institute, University of California, Berkeley, CA 94720; and lKavli Energy Nanosciences Institute, University of California, Berkeley, CA 94720 Edited by Feng Zhang, Broad Institute, Cambridge, MA, and approved July 22, 2019 (received for review May 27, 2019) Hydrophobic base stacking