Characterization of Two Nima Interacting Proteins Suggests
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeted Kinase Inhibitor Profiling Using a Hybrid Quadrupole-Orbitrap Mass Spectrometer
Targeted Kinase Inhibitor Profiling Note Application Using a Hybrid Quadrupole-Orbitrap Mass Spectrometer 1 2 3 Scott Peterman , Ryan D. Bomgarden , Rosa Viner 574 1Thermo Fisher Scientific, Cambridge, MA;2 Thermo Fisher Scientific, Rockford, IL; 3Thermo Fisher Scientific, San Jose, CA Key Words An emerging technology for identifying kinase inhibitor Q Exactive, targeted peptide quantification, msxSIM, kinome profiling by targets is based on chemical proteomic profiling of kinase MS, desthiobiotin nucleotide probes inhibitor specificity and binding affinity. This technology combines mass spectrometry (MS)-based identification Goal and quantitation with small molecule probe binding and To identify and quantify kinase inhibition by staurosporine using kinase enrichment to determine kinase active site occupancy active sites probes in combination with targeted, multiplexed SIM (msxSIM). during inhibition. One of these methods uses novel biotinylated ATP and/or ADP probes that irreversibly react with conserved lysine residues of kinase ATP binding Introduction sites.1,2 Selective enrichment of active-site peptides from Protein kinases are key enzymes involved in a wide array labeled kinase digests dramatically reduces background of complex cellular functions and pathways. Misregulation matrix and increases signal for MS analysis of low- or mutation of protein kinases underlies numerous disease abundance kinase peptides. Using this method, more than states, including tumorigenesis, making them ideal candidates 400 different protein and lipid kinases from various for drug development. However, identifying specific mammalian tissues and cell lines have been identified and kinase inhibitors is challenging due to the high degree of functionally assayed using targeted acquisition on an ion homology among subfamily members of the 500+ human trap mass spectrometer.1 The assays are available kinases. -

Nercc1, a Mammalian NIMA-Family Kinase, Binds the Ran Gtpase and Regulates Mitotic Progression
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Nercc1, a mammalian NIMA-family kinase, binds the Ran GTPase and regulates mitotic progression Joan Roig, Alexei Mikhailov, Christopher Belham, and Joseph Avruch1 Department of Molecular Biology and the Diabetes Unit and Medical Services, Massachusetts General Hospital, and the Department of Medicine, Harvard Medical School, Boston, Massachusetts 02114, USA The protein kinase NIMA is an indispensable pleiotropic regulator of mitotic progression in Aspergillus. Although several mammalian NIMA-like kinases (Neks) are known, none appears to have the broad importance for mitotic regulation attributed to NIMA. Nercc1 is a new NIMA-like kinase that regulates chromosome alignment and segregation in mitosis. Its NIMA-like catalytic domain is followed by a noncatalytic tail containing seven repeats homologous to those of the Ran GEF, RCC1, a Ser/Thr/Pro-rich segment, and a coiled-coil domain. Nercc1 binds to another NIMA-like kinase, Nek6, and also binds specifically to the Ran GTPase through both its catalytic and its RCC1-like domains, preferring RanGDP in vivo. Nercc1 exists as a homooligomer and can autoactivate in vitro by autophosphorylation. Nercc1 is a cytoplasmic protein that is activated during mitosis and is avidly phosphorylated by active p34Cdc2. Microinjection of anti-Nercc1 antibodies in prophase results in spindle abnormalities and/or chromosomal misalignment. In Ptk2 cells the outcome is prometaphase arrest or aberrant chromosome segregation and aneuploidy, whereas in CFPAC-1 cells prolonged arrest in prometaphase is the usual response. Nercc1 and its partner Nek6 represent a new signaling pathway that regulates mitotic progression. -

Biallelic Loss of Function NEK3 Mutations Deacetylate Α-Tubulin and Downregulate NUP205 That Predispose Individuals to Cilia-Re
Zhang et al. Cell Death and Disease (2020) 11:1005 https://doi.org/10.1038/s41419-020-03214-1 Cell Death & Disease ARTICLE Open Access Biallelic loss of function NEK3 mutations deacetylate α-tubulin and downregulate NUP205 that predispose individuals to cilia-related abnormal cardiac left–right patterning Yuan Zhang1, Weicheng Chen2, Weijia Zeng3, Zhouping Lu4 and Xiangyu Zhou4 Abstract Defective left–right (LR) organization involving abnormalities in cilia ultrastructure causes laterality disorders including situs inversus (SI) and heterotaxy (Htx) with the prevalence approximately 1/10,000 births. In this study, we describe two unrelated family trios with abnormal cardiac LR patterning. Through whole-exome sequencing (WES), we identified compound heterozygous mutations (c.805-1G >C; p. Ile269GlnfsTer8/c.1117dupA; p.Thr373AsnfsTer19) (c.29T>C; p.Ile10Thr/c.356A>G; p.His119Arg) of NEK3, encoding a NIMA (never in mitosis A)-related kinase, in two affected individuals, respectively. Protein levels of NEK3 were abrogated in Patient-1 with biallelic loss-of function (LoF) NEK3 mutations that causes premature stop codon. Subsequence transcriptome analysis revealed that NNMT (nicotinamide N-methyltransferase) and SIRT2 (sirtuin2) was upregulated by NEK3 knockdown in human retinal pigment epithelial (RPE) cells in vitro, which associates α-tubulin deacetylation by western blot and immunofluorescence. Transmission electron microscopy (TEM) analysis further identified defective ciliary ultrastructure 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; in Patient-1. Furthermore, inner ring components of nuclear pore complex (NPC) including nucleoporin (NUP)205, NUP188, and NUP155 were significantly downregulated in NEK3-silenced cells. In conclusion, we identified biallelic mutations of NEK3 predispose individual to abnormal cardiac left–right patterning via SIRT2-mediated α-tubulin deacetylation and downregulation of inner ring nucleoporins. -

Inhibition of ERK 1/2 Kinases Prevents Tendon Matrix Breakdown Ulrich Blache1,2,3, Stefania L
www.nature.com/scientificreports OPEN Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown Ulrich Blache1,2,3, Stefania L. Wunderli1,2,3, Amro A. Hussien1,2, Tino Stauber1,2, Gabriel Flückiger1,2, Maja Bollhalder1,2, Barbara Niederöst1,2, Sandro F. Fucentese1 & Jess G. Snedeker1,2* Tendon extracellular matrix (ECM) mechanical unloading results in tissue degradation and breakdown, with niche-dependent cellular stress directing proteolytic degradation of tendon. Here, we show that the extracellular-signal regulated kinase (ERK) pathway is central in tendon degradation of load-deprived tissue explants. We show that ERK 1/2 are highly phosphorylated in mechanically unloaded tendon fascicles in a vascular niche-dependent manner. Pharmacological inhibition of ERK 1/2 abolishes the induction of ECM catabolic gene expression (MMPs) and fully prevents loss of mechanical properties. Moreover, ERK 1/2 inhibition in unloaded tendon fascicles suppresses features of pathological tissue remodeling such as collagen type 3 matrix switch and the induction of the pro-fbrotic cytokine interleukin 11. This work demonstrates ERK signaling as a central checkpoint to trigger tendon matrix degradation and remodeling using load-deprived tissue explants. Tendon is a musculoskeletal tissue that transmits muscle force to bone. To accomplish its biomechanical function, tendon tissues adopt a specialized extracellular matrix (ECM) structure1. Te load-bearing tendon compart- ment consists of highly aligned collagen-rich fascicles that are interspersed with tendon stromal cells. Tendon is a mechanosensitive tissue whereby physiological mechanical loading is vital for maintaining tendon archi- tecture and homeostasis2. Mechanical unloading of the tissue, for instance following tendon rupture or more localized micro trauma, leads to proteolytic breakdown of the tissue with severe deterioration of both structural and mechanical properties3–5. -

Checking Neks: Overcoming a Bottleneck in Human Diseases
molecules Review Checking NEKs: Overcoming a Bottleneck in Human Diseases 1, 1,2, 1,2,3, Andressa Peres de Oliveira y, Luidy Kazuo Issayama y, Isadora Carolina Betim Pavan y, Fernando Riback Silva 1,2, Talita Diniz Melo-Hanchuk 1,2, Fernando Moreira Simabuco 3 and Jörg Kobarg 2,* 1 Instituto de Biologia, Departamento de Bioquímica e Biologia Tecidual, Universidade Estadual de Campinas, Campinas, São Paulo 13083-862, Brazil; [email protected] (A.P.d.O.); [email protected] (L.K.I.); [email protected] (I.C.B.P.); [email protected] (F.R.S.); [email protected] (T.D.M.-H.) 2 Faculdade de Ciências Farmacêuticas, Universidade Estadual de Campinas, Campinas, São Paulo 13083-871, Brazil 3 Laboratório Multidisciplinar em Alimentos e Saúde, Faculdade de Ciências Aplicadas, Universidade Estadual de Campinas, São Paulo 13484-350, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-19-3521-8143 These authors contributed equally to this work. y Received: 10 March 2020; Accepted: 9 April 2020; Published: 13 April 2020 Abstract: In previous years, several kinases, such as phosphoinositide 3-kinase (PI3K), mammalian target of rapamycin (mTOR), and extracellular-signal-regulated kinase (ERK), have been linked to important human diseases, although some kinase families remain neglected in terms of research, hiding their relevance to therapeutic approaches. Here, a review regarding the NEK family is presented, shedding light on important information related to NEKs and human diseases. NEKs are a large group of homologous kinases with related functions and structures that participate in several cellular processes such as the cell cycle, cell division, cilia formation, and the DNA damage response. -

Comprehensive Substrate Specificity Profiling of the Human
bioRxiv preprint doi: https://doi.org/10.1101/515221; this version posted January 8, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 2 3 4 5 Comprehensive Substrate Specificity Profiling of the 6 Human Nek Kinome Reveals Unexpected Signaling Outputs 7 8 9 Bert van de Kooij1, Pau Creixell1, Anne van Vlimmeren1, Brian A. Joughin1, 10 Chad J. Miller2,*, Nasir Haider3, Rune Linding4, Vuk Stambolic3, 11 Benjamin E. Turk2, Michael B. Yaffe1,5,6 12 13 14 15 16 17 18 19 20 21 22 23 24 1. Koch Institute for Integrative Cancer Research, MIT Center for Precision Cancer Medicine, 25 Departments of Biology and Bioengineering, Massachusetts Institute of Technology, 26 Cambridge, MA, USA. 27 2. Department of Pharmacology, Yale School of Medicine, New Haven, CT, USA 28 3. Department of Medical Biophysics, University of Toronto, Toronto, Canada 29 4. Biotech Research and Innovation Center, Faculty of Health and Medical Sciences, University 30 of Copenhagen, Copenhagen, Denmark 31 5. Department of Surgery, Beth Israel Deaconess Medical Center, Divisions of Acute Care 32 Surgery, Trauma, and Critical Care and Surgical Oncology, Harvard Medical School, Boston, 33 USA. 34 6. To whom correspondence should be addressed. E-mail [email protected] 35 * Current address: Department of Biochemistry, University of Washington, Seattle, WA, USA bioRxiv preprint doi: https://doi.org/10.1101/515221; this version posted January 8, 2019. -

Intracellular Calcium Release Modulates Polycystin-2 Trafficking Ayako Miyakawa1,2, Cristián Ibarra1, Seth Malmersjö1, Anita Aperia3, Peter Wiklund2 and Per Uhlén1*
Miyakawa et al. BMC Nephrology 2013, 14:34 http://www.biomedcentral.com/1471-2369/14/34 RESEARCH ARTICLE Open Access Intracellular calcium release modulates polycystin-2 trafficking Ayako Miyakawa1,2, Cristián Ibarra1, Seth Malmersjö1, Anita Aperia3, Peter Wiklund2 and Per Uhlén1* Abstract Background: Polycystin-2 (PC2), encoded by the gene that is mutated in autosomal dominant polycystic kidney disease (ADPKD), functions as a calcium (Ca2+) permeable ion channel. Considerable controversy remains regarding the subcellular localization and signaling function of PC2 in kidney cells. Methods: We investigated the subcellular PC2 localization by immunocytochemistry and confocal microscopy in primary cultures of human and rat proximal tubule cells after stimulating cytosolic Ca2+ signaling. Plasma membrane (PM) Ca2+ permeability was evaluated by Fura-2 manganese quenching using time-lapse fluorescence microscopy. Results: We demonstrated that PC2 exhibits a dynamic subcellular localization pattern. In unstimulated human or rat proximal tubule cells, PC2 exhibited a cytosolic/reticular distribution. Treatments with agents that in various ways affect the Ca2+ signaling machinery, those being ATP, bradykinin, ionomycin, CPA or thapsigargin, resulted in increased PC2 immunostaining in the PM. Exposing cells to the steroid hormone ouabain, known to trigger Ca2+ oscillations in kidney cells, caused increased PC2 in the PM and increased PM Ca2+ permeability. Intracellular Ca2+ buffering with BAPTA, 2+ inositol 1,4,5-trisphosphate receptor (InsP3R) inhibition with 2-aminoethoxydiphenyl borate (2-APB) or Ca /Calmodulin- dependent kinase inhibition with KN-93 completely abolished ouabain-stimulated PC2 translocation to the PM. Conclusions: These novel findings demonstrate intracellular Ca2+-dependent PC2 trafficking in human and rat kidney cells, which may provide new insight into cyst formations in ADPKD. -

Kinome Expression Profiling to Target New Therapeutic Avenues in Multiple Myeloma
Plasma Cell DIsorders SUPPLEMENTARY APPENDIX Kinome expression profiling to target new therapeutic avenues in multiple myeloma Hugues de Boussac, 1 Angélique Bruyer, 1 Michel Jourdan, 1 Anke Maes, 2 Nicolas Robert, 3 Claire Gourzones, 1 Laure Vincent, 4 Anja Seckinger, 5,6 Guillaume Cartron, 4,7,8 Dirk Hose, 5,6 Elke De Bruyne, 2 Alboukadel Kassambara, 1 Philippe Pasero 1 and Jérôme Moreaux 1,3,8 1IGH, CNRS, Université de Montpellier, Montpellier, France; 2Department of Hematology and Immunology, Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium; 3CHU Montpellier, Laboratory for Monitoring Innovative Therapies, Department of Biologi - cal Hematology, Montpellier, France; 4CHU Montpellier, Department of Clinical Hematology, Montpellier, France; 5Medizinische Klinik und Poliklinik V, Universitätsklinikum Heidelberg, Heidelberg, Germany; 6Nationales Centrum für Tumorerkrankungen, Heidelberg , Ger - many; 7Université de Montpellier, UMR CNRS 5235, Montpellier, France and 8 Université de Montpellier, UFR de Médecine, Montpel - lier, France ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2018.208306 Received: October 5, 2018. Accepted: July 5, 2019. Pre-published: July 9, 2019. Correspondence: JEROME MOREAUX - [email protected] Supplementary experiment procedures Kinome Index A list of 661 genes of kinases or kinases related have been extracted from literature9, and challenged in the HM cohort for OS prognostic values The prognostic value of each of the genes was computed using maximally selected rank test from R package MaxStat. After Benjamini Hochberg multiple testing correction a list of 104 significant prognostic genes has been extracted. This second list has then been challenged for similar prognosis value in the UAMS-TT2 validation cohort. -

© Copyright 2016 Antonio Abeyta
© Copyright 2016 Antonio Abeyta A High Throughput RNAi Screen Reveals the Protein Kinase NEK8 Regulates Replication Fork Protection via the DNA Repair Protein RAD51 Antonio Abeyta A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2016 Reading Committee: Toshiyasu Taniguchi, Chair Edith Wang Gerald Smith Program Authorized to Offer Degree: Molecular and Cellular Biology University of Washington Abstract A High Throughput RNAi Screen Reveals the Protein Kinase NEK8 Regulates Replication Fork Protection via the DNA Repair Protein RAD51 Antonio Abeyta Chair of the Supervisory Committee: Affiliate Associate Professor Toshiyasu Taniguchi Department of Pathology Proteins essential for homologous recombination play a pivotal role in the repair of DNA double strand breaks, DNA inter-strand crosslinks and replication fork stability. Defects in homologous recombination also play a critical role in the development of cancer and the sensitivity of these cancers to chemotherapy. RAD51, an essential factor for homologous recombination and replication fork protection, accumulates and forms immunocytochemically detectable nuclear foci at sites of DNA damage. To identify previously unknown kinases that may regulate RAD51 localization to sites of DNA damage, we performed a human kinome siRNA library screen, using DNA damage-induced RAD51 focus formation as readout. We found that NEK8, a NIMA family kinase member, is required for efficient DNA damage-induced RAD51 focus formation. Interestingly, we found that knockout of Nek8 in murine embryonic fibroblasts leads to cellular sensitivity to the replication inhibitor, hydroxyurea (HU), but not to other DNA damaging agents. Furthermore, we show that NEK8 is required for proper replication fork protection following replication stall with HU. -

Nanostring®: Product Data Sheet | Ncounter® GX Human Kinase
PRODUCT DATA SHEET nCounter® GX Human Kinase Kit nCounter® GX Human Kinase Kit Product Highlights Highly Curated • Our expert bio-informaticists use a very rigorous process in selecting the most meaningful set of genes Efficient • Multiplexed assay profiles 522 human kinase genes in a single reaction Cost-effective • Gold-standard data at a fraction of the cost Quick Turnaround Time • Complete kit with all consumables ready to ship next day nCounter® GX Human Kinase Kit The nCounter GX Human Kinase Kit is a comprehensive list of 522 human The nCounter Human Kinase Kit represents 99% of the KinBase content for genes known to be differentially expressed in the kinome. Human. With the nCounter GX Human Kinase Kit, scientists can leverage a pre-designed The final nCounter GX Human Kinase Kit consists of 522 protein kinase- panel to accelerate their research and quickly generate expression data for a related genes and 14 internal reference genes. For the gene list and additional large panel of protein kinase-related genes. information about this gene set, visit the nCounter Pre-built Panels product page at: www.nanostring.com. The gene list was compiled using the KinBase database at www.kinase.com. Home > Products > nCounter Gene Expression CodeSets > Pre-built Panels The database is based on the publication, The Protein Kinase Complement of the Human Genome, by G Manning, DB Whyte, R Martinez, T Hunter, S Sudarsanam (2002). Science 298:1912-1934. Molecules That Count® Translational Research Gene Expression miRNA Expression Copy Number Variation 1 PRODUCT DATA SHEET nCounter® GX Human Kinase Kit nCounter® Analysis System Overview The nCounter Analysis System from NanoString offers a cost-effective way to easily profile hundreds of gene transcripts simultaneously with high sensitivity and precision. -
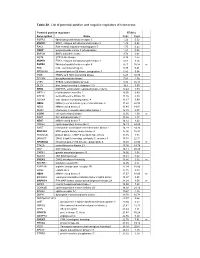
Table S1. List of Potential Positive and Negative Regulators of Telomerase
Table S1. List of potential positive and negative regulators of telomerase. Potential positive regulators RTA(%) Gene symbol Name Rep1 Rep2 FGFR3 fibroblast growth factor receptor 3 1.26 5.52 MAPK3 ERK1, mitogen activated protein kinase 3 1.39 0.25 RAC1 Ras-related, migration-inducing gene 5 1.75 0.22 PNKP polynucleotide kinase 3’-phosphatase 1.91 0.02 BMP2K BMP2 inducible kinase 3.74 0.01 MST4 STE20-like kinase 4.19 2.55 MAPK1 ERK2, mitogen activated protein kinase 1 4.63 8.54 FGFR4 fibrablast growth factor receptor 4 4.71 16.58 nr FES feline sarcoma oncogene 5.38 6.41 RPS6KA5 ribosomal protein S6 kinase, polypeptide 5 5.86 9.64 TNIK TRAF2 and NCK interacting kinase 6.27 10.38 DTYMK deoxythymidylate kinase 7.84 4.38 LYK5 STE20-related adaptor protein 8.92 16.38 DLG4 disc, large homolog 4, chapsyn-110 10.4 3.59 ERK8 MAPK15, extracellular regulated kinase 8 delta 10.44 3.59 MPZL1 myelin protein zero-like 1 10.95 0.49 STK19 serine/threonine kinase 19 11.16 4.33 ADCK4 aarF domain containing kinase 4 11.17 3.99 NEK2 NIMA (never in mitosis gene)-related kinase 2 11.22 22.58 NEK6 NIMA-related kinase 6 11.40 14.01 IRAK1 interleukin-1 receptor-associated kinase 1 12.15 0.07 CCRK cell cycle related kinase 12.56 4.30 nr PAK7 P21-activated kinase 7 13.46 1.17 NEK7 NIMA-related kinase 7 14.12 8.43 CDKL2 cyclin-dependent kinase-like 2 14.19 24.48 SAST microtubule associated serine/threonine kinase 1 14.29 34.36 MAP3K4 MAP protein kinase kinase kinase 4 15.00 10.92 PRKAR2B protein kinase, cAMP-dependent type II beta 15.16 1.35 DNAJC3 DNAJ (Hsp40) homolog, -
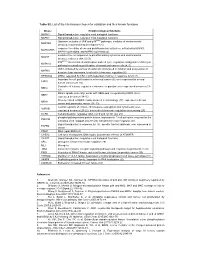
Table S2. List of the Hits Kinases Chosen for Validation and Their Known Functions
Table S2. List of the hits kinases chosen for validation and their known functions kinase Known biological functions MAPK3 Signal transduction, regulates most biological functions MAPK1 Signal transduction, regulates most biological functions Upstream activator of JNK and p38MAPK pathways, mediator of environmental MAP3K4 stresses; required during development (1) response to cellular stress and proinflammatory cytokines, activated by MAPK1, MAPKAPK5 MAPK14/p38-alpha, and MAPK11/p38-beta (2) mediates the cell responses to proinflammatory cytokines and environmental MA2K7 stresses; activates JNK1/2 (3) P38MAPKα stress related transcription and cell cycle regulation; antagonist of JNK/c-jun MAPK14 pathway in controlling proliferation of normal and cancer cells (4, 5) JNK1, activated by various stressful stimuli involved in initiation and progression of MAPK8 hepatocellular carcinoma. Involved in telomerase regulation (6) RPS6KA4 MSK2; regulated by CK2 in p38-dependent manner in response to UV (7) Important for cell proliferation in colorectal cancer (8); over-expressed in several ERK8 human cancers (9, 10) Controller of mitosis, regulates centrosome separation; over-expressed in cancer (11- NEK2 13) Mitotic spindle assembly; works with NEK6 and is regulated by NEK9. Over- NEK7 expressed in cancer (14-16) Closely related to NEK9; mainly involved in cilia biology (17); expressed in breast NEK8 cancer and pancreatic cancer (18, 19) Central regulator of mitosis, chromosome segregation and cytokinesis; over- AURKB expressed in cancer (20-22). Involved in telomerase regulation via surviving (23) CCRK Cell proliferation, regulates cdk2, cyc E and cyc D1 (24, 25) phospholipid-dependent protein kinase, important for T-cell activation; required for the PRKCQ activation of NF-kappaB and AP (26).