Evidence of Postzygotic Mosaicism in a Transmitted Form of Conradi-Hu¨ Nermann-Happle Syndrome Associated with a Novel EBP Mutation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
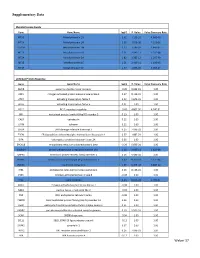
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
Weber 37 Supplementary Data
Supplementary Data Metallothionein Family Gene Gene Name logFC P. Value False Discovery Rate MT1G Metallothionein 1G 2.62 1.19E-10 4.66E-08 MT1H Metallothionein 1H 2.58 4.87E-08 7.12E-06 MT1M Metallothionein 1M 2.23 1.18E-07 1.44E-05 MT1X Metallothionein 1X 2.21 2.64E-11 1.25E-08 MT2A Metallothionein 2A 1.82 1.38E-12 1.26E-09 MT1E Metallothionein 1E 1.51 2.41E-11 1.18E-08 MT1F Metallothionein 1F 1.47 1.85E-09 4.09E-07 Unfolded Protein Response Gene Gene Name logFC P. Value False Discovery Rate AMFR autocrine motility factor receptor -0.20 9.94E-01 1.00 ASK1 mitogen-activated protein kinase kinase kinase 5 0.27 8.11E-01 1.00 ATF4 activating transcription factor 4 0.22 9.67E-01 1.00 ATF6 activating transcription factor 6 0.01 1.00 1.00 BCL2 BCL2, apoptosis regulator -0.43 4.80E-02 4.24E-01 BIP heat shock protein family A (Hsp70) member 5 0.13 1.00 1.00 CALR calreticulin 0.12 1.00 1.00 CANX calnexin 0.12 1.00 1.00 CHOP DNA damage inducible transcript 3 0.25 7.99E-01 1.00 EDEM ER degradation enhancing alpha-mannosidase like protein 1 0.37 1.88E-01 1.00 EIFA eukaryotic translation initiation factor 2A 0.06 1.00 1.00 ERO1LB endoplasmic reticulum oxidoreductase 1 beta -0.29 5.69E-01 1.00 GADD34 protein phosphatase 1 regulatory subunit 15A 1.18 3.66E-11 1.62E-08 GRP94 heat shock protein 90 beta family member 1 0.16 9.98E-01 1.00 HSPH1 heat shock protein family H (Hsp110) member 1 0.62 9.70E-06 5.12E-04 INSIG1 insulin induced gene 1 1.69 8.47E-18 3.88E-14 IRE1 endoplasmic reticulum to nucleus signaling 1 0.26 8.13E-01 1.00 JNK1 mitogen-activated -

Supplement 1A Steffensen Et
Liver Wild-type Knockout C T C T 1 2 4 7 8 9 1 2 4 5 7 9 1 1 1 1 1 1 C C C T T T C C C T T T W W W W W W K K K K K K IMAGE:793166 RIKEN cDNA 6720463E02 gene IMAGE:1447421 ESTs, Weakly similar to ZF37 MOUSE ZINC FINGER PROTEIN 37 [M.musculus] IMAGE:934291 RIKEN cDNA 2810418N01 gene IMAGE:1247525 small EDRK-rich f2actor IMAGE:1449402 expressed sequence AW321064 IMAGE:1279847 ESTs IMAGE:518737 expressed sequence AW049941 IMAGE:860231 a disintegrin and metalloproteinase domain 17 IMAGE:642836 CD86 antigen IMAGE:1003885 phosphoribosyl pyrophosphate sy1nthetase IMAGE:524862 RIKEN cDNA 5730469D23 gene IMAGE:1264473 protein inhibitor of activat1ed STAT IMAGE:847035 RIKEN cDNA 4833422F06 gene IMAGE:374550 requiem IMAGE:976520 nuclear receptor coact4ivator IMAGE:1264311 Unknown IMAGE:976735 expressed sequence AI987692 IMAGE:976659 cathepsLin IMAGE:1477580 RIKEN cDNA 1600010J02 gene IMAGE:1277168 ribosomal protein, large, P1 IMAGE:524842 RIKEN cDNA 0710008D09 gene IMAGE:373019 split hand/foot delete1d gene IMAGE:404428 expressed sequence AI413851 IMAGE:619810 RIKEN cDNA 1700003F10 gene IMAGE:1749558 caspase 3, apoptosis related cysteine protease IMAGE:718718 RIKEN cDNA 2810003F23 gene IMAGE:819789 Unknown IMAGE:524474 ATP-binding cassette, sub-family A ABC1, member IMAGE:804950 Mus musculus, Similar to ribosomal protein S20, clone MGC:6876 IMAGE:2651405, mRNA, complete cds IMAGE:806143 gap junction membrane channel prot2ein beta IMAGE:1745887 expressed sequence AI836376 IMAGE:779426 RIKEN cDNA 5230400G24 gene IMAGE:1125615 Unknown IMAGE:535025 DNA -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -
Figures Intermediary Ontogeny.Pptx
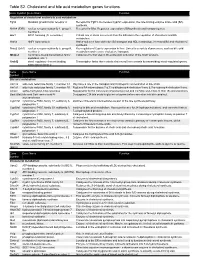
Table S2. Cholesterol and bile acid metabolism genes functions. Gene Symbol Gene Name Function Regulation of cholesterol and/or bile acid metabolism Fgfr4 fibroblast growth factor receptor 4 Receptor for Fgf15. Decreases Cyp7a1 expression, the rate-limiting enzyme in bile acid (BA) synthesis. Nr1h4 (FXR) nuclear receptor subfamily 1, group H, Receptor for BAs. Regulates expression of BAsynthesis and transport genes. member 4 Arv1 ARV1 homolog (S. cerevisiae) Critical role in sterol movement from the ER and in the regulation of cholesterol and BA metabolism. Hnf1a HNF1 homeobox A Hnf1a-null mice have defective BA transport and HDL metabolism, increased BA and cholesterol synthesis. Nr5a2 (Lrh1) nuclear receptor subfamily 5, group A, Key regulator of Cyp7a expression in liver. Linked to a variety of processes, such as bile acid member 2 metabolism and reverse cholesterol transport. Mbtps1 membrane-bound transcription factor Catalyzes the first step in the proteolytic activation of the Srebf proteins. peptidase, site 1 Srebf2 sterol regulatory element binding Transcription factor that controls cholesterol homeostasis by transcribing sterol-regulated genes. transcription factor 2 Gene Gene Name Function Symbol Bile acid metabolism Akr1c6 aldo-keto reductase family 1, member C1 May have a role in the transport and intrahepatic concentration of bile acids. Akr1d1 aldo-keto reductase family 1, member D1 Reduces BA intermediates 7-α,12-α-dihydroxy-4-cholesten-3-one & 7-α-hydroxy-4-cholesten-3-one. Amacr alpha-methylacyl-CoA racemase Responsible for the conversion of pristanoyl-CoA and C27-bile acyl-CoAs to their (S)-stereoisomers. Baat (Bat) bile acid CoA: amino acid N- Conjugates C24 bile acids to glycine or taurine before excretion into bile canaliculi. -
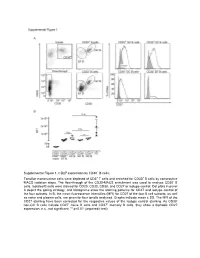
Supplemental Figure 1. CD27 Expression by CD30+ B Cells
Supplemental Figure 1. CD27 expression by CD30+ B cells. Tonsillar mononuclear cells were depleted of CD3+ T cells and enriched for CD30+ B cells by consecutive MACS isolation steps. The flow-through of the CD30-MACS enrichment was used to analyze CD30- B cells. Isolated B cells were stained for CD20, CD30, CD38, and CD27 or isotype control. Dot plots in panel A depict the gating strategy, and histograms show the staining patterns for CD27 and isotype control of the four subsets. In B, the mean fluorescence intensities (MFI) for CD27 of the four B cell subsets, as well as naive and plasma cells, are given for four tonsils analyzed. Graphs indicate mean ± SD. The MFI of the CD27 staining have been corrected for the respective values of the isotype control staining. As CD30- non-GC B cells include CD27- naïve B cells and CD27+ memory B cells, they show a biphasic CD27 expression. n.s., not significant; ** p<0.01 (unpaired t test). Supplemental Figure 2. Interleukin receptor expression by CD30+ B cells and relatedness of CD30+ EF B cells to CD21low B cells. mRNA and surface protein expression of IL2RB (A), IL21R (B) and CD21 (C) are shown for indicated B cell subsets of five tonsils (conventional (conv.) CD30- GC (germinal center) B cells (CD20highCD38+), CD30+ GC B cells, CD30- memory and CD30+ EF (extra follicular) B cells (CD20+CD38-/lowCD27+), and plasma cells (PC) (CD20+CD38high). mRNA data originate from Affymetrix genechip analyses (Tiacci et al., Blood, 2012; 120:4609-4620) and were analyzed using an unpaired t test. -

Supplemental Figure 1. Protein-Protein Interaction Network with Increased Expression in Fteb During the Luteal Phase
Supplemental Figure 1. Protein-protein interaction network with increased expression in FTEb during the luteal phase. Supplemental Figure 2. Protein-protein interaction network with decreased expression in FTEb during luteal phase. LEGENDS TO SUPPLEMENTAL FIGURES Supplemental Figure 1. Protein-protein interaction network with increased expression in FTEb during the luteal phase. Submission of probe sets differentially expressed in the FTEb specimens that clustered with SerCa as well as those specifically altered in FTEb luteal samples to the online I2D database revealed overlapping networks of proteins with increased expression in the four FTEb samples and/or FTEb luteal samples overall. Proteins are represented by nodes, and known and predicted first-degree interactions are represented by solid lines. Genes encoding proteins shown as large ovals highlighted in blue were exclusively found in the first comparison (Manuscript Figure 2), whereas those highlighted in red were only found in the second comparison (Manuscript Figure 3). Genes encoding proteins shown as large ovals highlighted in black were found in both comparisons. The color of each node indicates the ontology of the corresponding protein as determined by the Online Predicted Human Interaction Database (OPHID) link with the NAViGaTOR software. Supplemental Figure 2. Protein-protein interaction network with decreased expression in FTEb during the luteal phase. Submission of probe sets differentially expressed in the FTEb specimens that clustered with SerCa as well as those specifically altered in FTEb luteal samples to the online I2D database revealed overlapping networks of proteins with decreased expression in the four FTEb samples and/or FTEb luteal samples overall. Proteins are represented by nodes, and known and predicted first-degree interactions are represented by solid lines. -

Transcriptional Regulators Are Upregulated in the Substantia Nigra
Journal of Emerging Investigators Transcriptional Regulators are Upregulated in the Substantia Nigra of Parkinson’s Disease Patients Marianne Cowherd1 and Inhan Lee2 1Community High School, Ann Arbor, MI 2miRcore, Ann Arbor, MI Summary neurological conditions is an established practice (3). Parkinson’s disease (PD) affects approximately 10 Significant gene expression dysregulation in the SN and million people worldwide with tremors, bradykinesia, in the striatum has been described, particularly decreased apathy, memory loss, and language issues. Though such expression in PD synapses. Protein degradation has symptoms are due to the loss of the substantia nigra (SN) been found to be upregulated (4). Mutations in SNCA brain region, the ultimate causes and complete pathology are unknown. To understand the global gene expression (5), LRRK2 (6), and GBA (6) have also been identified changes in SN, microarray expression data from the SN as familial markers of PD. SNCA encodes alpha- tissue of 9 controls and 16 PD patients were compared, synuclein, a protein found in presynaptic terminals that and significantly upregulated and downregulated may regulate vesicle presence and dopamine release. genes were identified. Among the upregulated genes, Eighteen SNCA mutations have been associated with a network of 33 interacting genes centered around the PD and, although the exact pathogenic mechanism is cAMP-response element binding protein (CREBBP) was not confirmed, mutated alpha-synuclein is the major found. The downstream effects of increased CREBBP- component of protein aggregates, called Lewy bodies, related transcription and the resulting protein levels that are often found in PD brains and may contribute may result in PD symptoms, making CREBBP a potential therapeutic target due to its central role in the interactive to cell death. -

Supplemental Data
Supplementary Table 1. Gene sets from Figure 6. Lists of genes from each individual gene set defined in Figure 6, including the fold-change in expression of each gene in treatment group pair-wise comparisons. ENSEMBL: Ensembl gene identifier; Symbol: official gene symbol; logFC: log fold change; p value: significance of fold-change in a pair-wise comparison, P<0.05 cut-off; FDR: false discovery rate, expected proportion of false positives among the differentially expressed genes in a pair-wise comparison (FDR<0.25 cut-off). Sup. Table 1 SET I CP versus Sal CP versus CP+DCA DCA versus Sal ENSEMBL Symbol logFC PValue FDR logFC PValue FDR logFC PValue FDR Desc ENSMUSG00000020326 Ccng1 2.64 0.00 0.00 -0.06 0.13 0.96 0.40 0.00 0.23 cyclin G1 [Source:MGI Symbol;Acc:MGI:102890] ENSMUSG00000031886 Ces2e 3.97 0.00 0.00 -0.24 0.02 0.28 0.01 1.00 1.00 carboxylesterase 2E [Source:MGI Symbol;Acc:MGI:2443170] ENSMUSG00000041959 S100a10 2.31 0.00 0.00 -0.21 0.02 0.23 -0.11 0.53 1.00 S100 calcium binding protein A10 (calpactin) [Source:MGI Symbol;Acc:MGI:1339468] ENSMUSG00000092341 Malat1 1.09 0.00 0.00 -0.11 0.20 1.00 0.66 0.00 0.00 metastasis associated lung adenocarcinoma transcript 1 (non-coding RNA) [Source:MGI Symbol;Acc:MGI:1919539] ENSMUSG00000072949 Acot1 1.73 0.00 0.00 -0.22 0.01 0.12 -0.44 0.01 1.00 acyl-CoA thioesterase 1 [Source:MGI Symbol;Acc:MGI:1349396] ENSMUSG00000064339 mt-Rnr2 1.09 0.00 0.00 -0.08 0.17 1.00 0.67 0.00 0.07 mitochondrially encoded 16S rRNA [Source:MGI Symbol;Acc:MGI:102492] ENSMUSG00000025934 Gsta3 1.86 0.00 0.00 -0.28 -

Rieck Et Al.- Supplementary Gene List Figure 1- FINAL
Separation of GFP+ and GFP- populations from Mip-GFP mice. (A) SSC (side scatter) is proportional to cell complexity and depends on the number of organelles inside the cell. FSC (forward scatter) correlates with cell size. GFP 530/30-A measures strength of an individual cells GFP intensity. (A-D) Freshly isolated islets from CD1+ female mouse (no GFP expression) were used to calibrate the cell sorter before GFP sorting began. (E) Plotting both SSC and FSC shows all cells collected as shown by the highlighted gate. (F) Only singlet or non-aggregated cells were used for ecient cell sorting. (G-H) Two distinct populations of cells were collected separately for further analysis. Dierentially expressed genes during pregnancy day 14.5 (FC 1.5 and FDR 20%) GeneSymbol Fold Change GeneName C85627 53.11 expressed sequence C85627 Cldn8 51.06 claudin 8 LOC382109 27.98 Hypothetical protein E330023O18 Lrrc55 26.70 leucine rich repeat containing 55 Fbxw14 25.85 F-box and WD-40 domain protein 14 Tph1 19.14 tryptophan hydroxylase 1 Itk 18.16 IL2-inducible T-cell kinase Gdf3 17.91 growth differentiation factor 3 Tph2 12.09 tryptophan hydroxylase 2 Cish 10.88 cytokine inducible SH2-containing protein 8.91 EG382156 8.77 predicted gene, EG382156 1700124P09Rik 8.70 RIKEN cDNA 1700124P09 gene Gpc6 7.29 glypican 6 5830443L24Rik 7.25 RIKEN cDNA 5830443L24 gene Fmo1 7.20 flavin containing monooxygenase 1 5830417I10Rik 7.09 RIKEN cDNA 5830417I10 gene Sftpd 6.98 surfactant associated protein D 6430550H21Rik 6.39 RIKEN cDNA 6430550H21 gene 1810009J06Rik 6.38 RIKEN -
Potential Role of Increased Oxygenation in Altering Perinatal Adrenal Steroidogenesis
nature publishing group Articles Basic Science Investigation Potential role of increased oxygenation in altering perinatal adrenal steroidogenesis Vishal Agrawal1, Meng Kian Tee1, Jie Qiao1, Marcus O. Muench2,3 and Walter L. Miller1 BACKGROUND: At birth, the large fetal adrenal involutes rap- respond to adrenocorticotropic hormone (1). The fetal adrenal idly, and the patterns of steroidogenesis change dramatically; transiently expresses 3β-hydroxysteroid dehydrogenase, type the event(s) triggering these changes remain largely unex- 2 (3βHSD2, encoded by HSD3B2) at about 8–10 wk, permit- plored. Fetal abdominal viscera receive hypoxic blood having a ting fetal adrenal cortisol synthesis at the same time when male partial pressure of oxygen of only ~2 kPa (20–23 mm Hg); peri- genital development occurs, thus helping to prevent the viril- natal circulatory changes change this to adult values (~20 kPa). ization of female fetuses by suppressing fetal adrenal andro- We hypothesized that transition from fetal hypoxia to postna- gen synthesis (1). The fetal adrenal has relatively little 3βHSD2 tal normoxia participates in altering perinatal steroidogenesis. activity after 12 wk (1,2) but has 17α-hydroxylase and robust METHODS: We grew midgestation human fetal adrenal cells 17,20 lyase activity (both catalyzed by cytochrome P450c17, and human NCI-H295A adrenocortical carcinoma cells in 2% encoded by CYP17A1), considerable sulfotransferase activity, O2, then transitioned them to 20% O2 and quantitated ste- and little steroid sulfatase activity accounting for its abun- roidogenic mRNAs by quantitative PCR and microarrays. dant production of dehydroepiandrosterone (DHEA) and its RESULTS: Transitioning fetal adrenal cells from hypoxia to sulfate (DHEAS). DHEAS is secreted, 16α-hydroxylated in normoxia increased mRNAs for 17α-hydroxylase/17,20 lyase the fetal liver by CYP3A7 (3–5), and then acted on by pla- (P450c17), 3β-hydroxysteroid dehydrogenase (3βHSD2), and cental 3βHSD1, 17βHSD1 (17β-hydroxysteroid dehydroge- steroidogenic acute regulatory protein (StAR). -

Emopamil Binding Protein (EBP) (NM 006579) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP301706 emopamil binding protein (EBP) (NM_006579) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human emopamil binding protein (sterol isomerase) (EBP) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 26.2 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_006570 Locus ID: 10682 UniProt ID: Q15125, A0A024QYX0 RefSeq Size: 1191 Cytogenetics: Xp11.23 RefSeq ORF: 690 Synonyms: CDPX2; CHO2; CPX; CPXD; MEND This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 emopamil binding protein (EBP) (NM_006579) Human Recombinant Protein – TP301706 Summary: The protein encoded by this gene is an integral membrane protein of the endoplasmic reticulum. It is a high affinity binding protein for the antiischemic phenylalkylamine Ca2+ antagonist [3H]emopamil and the photoaffinity label [3H]azidopamil. It is similar to sigma receptors and may be a member of a superfamily of high affinity drug-binding proteins in the endoplasmic reticulum of different tissues.