Adasuve, INN-Loxapine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neurotransmitter Resource Guide
NEUROTRANSMITTER RESOURCE GUIDE Science + Insight doctorsdata.com Doctor’s Data, Inc. Neurotransmitter RESOURCE GUIDE Table of Contents Sample Report Sample Report ........................................................................................................................................................................... 1 Analyte Considerations Phenylethylamine (B-phenylethylamine or PEA) ................................................................................................. 1 Tyrosine .......................................................................................................................................................................................... 3 Tyramine ........................................................................................................................................................................................4 Dopamine .....................................................................................................................................................................................6 3, 4-Dihydroxyphenylacetic Acid (DOPAC) ............................................................................................................... 7 3-Methoxytyramine (3-MT) ............................................................................................................................................... 9 Norepinephrine ........................................................................................................................................................................ -

THE BIOSYNTHESIS of SOME PHENOLIC ALKALOIDS a Thesis Submitted by GEOFFREY MELVILLE THOMAS for the Degree of DOCTOR of PHILOSOPH
/1 THE BIOSYNTHESIS OF SOME PHENOLIC ALKALOIDS a thesis submitted by GEOFFREY MELVILLE THOMAS for the degree of DOCTOR OF PHILOSOPHY of THE UNIVERSITY OF LONDON Imperial College, June 1963. London, S. W.7. ABSTRACT A brief review of the biosynthesis of alkaloids, other than those of the Amaryllidaceae and morphine groups, is given. The biosynthesis of these two groups is discussed more fully with particular reference to the evidence for the Barton and Cohen concept of phenol oxidation as a biogenetic mechanism. The incorporation of labelled phenolic precursors, derivatives of norbelladine, has been shown and by means of multiple labelled experiments incorporation as a whole, without degradation, has been proved. Other experiments described have thrown light on the earlier stages of biogenesis. The norlaudanosine derivative, (±) reticuline, has been shown to be incorporated into morphine, and an in vitro synthesis of thebaine from (±) reticuline using a radiochemical dilution method is de-scribed. I am deeply grateful to Professor D. H. R. Barton and Dr. G. W. Kirby for the privilege and pleasure of working under their supervision and for their great help in matters chemical and non-chemical. To the Salters Company I would like to express my sincere thanks for the award of a scholarship and for their interest during the tenure of it. My thanks are also due to Dr. D. W. Turner for advice on counting techniques, Mr. D. Aldrich and his staff for valuable technical assistance, Miss J. Cuckney for microanalyses, Mr. R.H. Young who grew the daffodils and poppies and to my many friends and co-workers at Imperial College. -
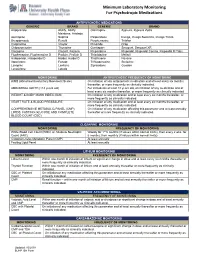
Minimum Laboratory Monitoring for Psychotropic Medications
Minimum Laboratory Monitoring For Psychotropic Medications ANTIPSYCHOTIC MEDICATIONS GENERIC BRAND GENERIC BRAND Aripiprazole Abilify, Abilify Olanzapine Zyprexa, Zyprexa Zydis Maintena, Aristada Asenapine Saphris Paliperidone Invega, Invega Sustenna, Invega Trinza Brexpiprazole Rexulti Perphenazine Trilafon Cariprazine Vraylar Pimozide Orap Chlorpromazine Thorazine Quetiapine Seroquel, Seroquel XR Clozapine Clozaril, Fazaclo Risperidone Risperdal, Risperdal Consta, Risperdal M Tabs Fluphenazine, Fluphenazine D Prolixin, Prolixin D Thioridazine Mellaril Haloperidol, Haloperidol D Haldol, Haldol D Thiothixene Navane Iloperidone Fanapt Trifluoperazine Stelazine Loxapine Loxitane Ziprasidone Geodon Lurasidone Latuda MONITORING ANTIPSYCHOTIC FREQUENCY OF MONITORING AIMS (Abnormal Involuntary Movement Scale) On initiation of any antipsychotic medication and at least every six months thereafter, or more frequently as clinically indicated. ABDOMINAL GIRTH (>18 years old) For individuals at least 18 years old, on initiation of any medication and at least every six months thereafter, or more frequently as clinically indicated. WEIGHT & BODY MASS INDEX (BMI) On initiation of any medication and at least every six months thereafter, or more frequently as clinically indicated. HEART RATE & BLOOD PRESSSURE On initiation of any medication and at least every six months thereafter, or more frequently as clinically indicated. COMPREHENSIVE METABOLIC PANEL (CMP), On initiation of any medication affecting this parameter and at least annually LIPIDS, FASTING -

Steven, Stacy (2012) the Pharmacology of the 5-HT2A Receptor and the Difficulty Surrounding Single Taret Models
Steven, Stacy (2012) The pharmacology of the 5-HT2A receptor and the difficulty surrounding single taret models. MSc(R) thesis. http://theses.gla.ac.uk/3636/ Copyright and moral rights for this thesis are retained by the author A copy can be downloaded for personal non-commercial research or study, without prior permission or charge This thesis cannot be reproduced or quoted extensively from without first obtaining permission in writing from the Author The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the Author When referring to this work, full bibliographic details including the author, title, awarding institution and date of the thesis must be given Glasgow Theses Service http://theses.gla.ac.uk/ [email protected] The pharmacology of the 5-HT2A receptor and the difficulty surrounding functional studies with single target models A thesis presented for the degree of Master of Science by research Stacy Steven April 2012 Treatment of many disorders can be frequently problematic due to the relatively non selective nature of many drugs available on the market. Symptoms can be complex and expansive, often leading to symptoms representing other disorders in addition to the primary reason for treatment. In particular mental health disorders fall prey to this situation. Targeting treatment can be difficult due to the implication of receptors in more than one disorder, and more than one receptor in a single disorder. In the instance of GPCRs, receptors such as the serotonin receptors (and in particular the 5-HT2A for the interest of this research) belong to a large family of receptors, the GPCR Class A super family. -

Schizophrenia Care Guide
August 2015 CCHCS/DHCS Care Guide: Schizophrenia SUMMARY DECISION SUPPORT PATIENT EDUCATION/SELF MANAGEMENT GOALS ALERTS Minimize frequency and severity of psychotic episodes Suicidal ideation or gestures Encourage medication adherence Abnormal movements Manage medication side effects Delusions Monitor as clinically appropriate Neuroleptic Malignant Syndrome Danger to self or others DIAGNOSTIC CRITERIA/EVALUATION (PER DSM V) 1. Rule out delirium or other medical illnesses mimicking schizophrenia (see page 5), medications or drugs of abuse causing psychosis (see page 6), other mental illness causes of psychosis, e.g., Bipolar Mania or Depression, Major Depression, PTSD, borderline personality disorder (see page 4). Ideas in patients (even odd ideas) that we disagree with can be learned and are therefore not necessarily signs of schizophrenia. Schizophrenia is a world-wide phenomenon that can occur in cultures with widely differing ideas. 2. Diagnosis is made based on the following: (Criteria A and B must be met) A. Two of the following symptoms/signs must be present over much of at least one month (unless treated), with a significant impact on social or occupational functioning, over at least a 6-month period of time: Delusions, Hallucinations, Disorganized Speech, Negative symptoms (social withdrawal, poverty of thought, etc.), severely disorganized or catatonic behavior. B. At least one of the symptoms/signs should be Delusions, Hallucinations, or Disorganized Speech. TREATMENT OPTIONS MEDICATIONS Informed consent for psychotropic -
![Selective Labeling of Serotonin Receptors Byd-[3H]Lysergic Acid](https://docslib.b-cdn.net/cover/9764/selective-labeling-of-serotonin-receptors-byd-3h-lysergic-acid-319764.webp)
Selective Labeling of Serotonin Receptors Byd-[3H]Lysergic Acid
Proc. Nati. Acad. Sci. USA Vol. 75, No. 12, pp. 5783-5787, December 1978 Biochemistry Selective labeling of serotonin receptors by d-[3H]lysergic acid diethylamide in calf caudate (ergots/hallucinogens/tryptamines/norepinephrine/dopamine) PATRICIA M. WHITAKER AND PHILIP SEEMAN* Department of Pharmacology, University of Toronto, Toronto, Canada M5S 1A8 Communicated by Philip Siekevltz, August 18,1978 ABSTRACT Since it was known that d-lysergic acid di- The objective in this present study was to improve the se- ethylamide (LSD) affected catecholaminergic as well as sero- lectivity of [3H]LSD for serotonin receptors, concomitantly toninergic neurons, the objective in this study was to enhance using other drugs to block a-adrenergic and dopamine receptors the selectivity of [3HJISD binding to serotonin receptors in vitro by using crude homogenates of calf caudate. In the presence of (cf. refs. 36-38). We then compared the potencies of various a combination of 50 nM each of phentolamine (adde to pre- drugs on this selective [3H]LSD binding and compared these clude the binding of [3HJLSD to a-adrenoceptors), apmo ie, data to those for the high-affinity binding of [3H]serotonin and spiperone (added to preclude the binding of [3H[LSD to (39). dopamine receptors), it was found by Scatchard analysis that the total number of 3H sites went down to 300 fmol/mg, compared to 1100 fmol/mg in the absence of the catechol- METHODS amine-blocking drugs. The IC50 values (concentrations to inhibit Preparation of Membranes. Calf brains were obtained fresh binding by 50%) for various drugs were tested on the binding of [3HLSD in the presence of 50 nM each of apomorphine (A), from the Canada Packers Hunisett plant (Toronto). -

Current P SYCHIATRY
Current p SYCHIATRY N ew Investigators Tips to manage and prevent discontinuation syndromes Informed tapering can protect patients when you stop a medication Sriram Ramaswamy, MD Shruti Malik, MBBS, MHSA Vijay Dewan, MD Instructor, department of psychiatry Foreign medical graduate Assistant professor Creighton University Department of psychiatry Omaha, NE University of Nebraska Medical Center Omaha, NE bruptly stopping common psychotropics New insights on psychotropic A —particularly antidepressants, benzodi- drug safety and side effects azepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with: This paper was among those entered in the 2005 • rebound or relapse of original symptoms Promising New Investigators competition sponsored • uncomfortable new physical and psycho- by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s competition logical symptoms was “New insights on psychotropic drug safety and • physiologic withdrawal at times. side effects.” To increase health professionals’ awareness of URRENT SYCHIATRY 1 C P is honored to publish this peer- the risk of these adverse effects, this article reviewed, evidence-based article on a clinically describes discontinuation syndromes associated important topic for practicing psychiatrists. with various psychotropics and offers strategies to NMSIS is dedicated to reducing morbidity and anticipate, recognize, and manage them. mortality of NMS by improving medical and psychiatric care of patients with heat-related disorders; providing -

(Xenazine); Deutetrabenazine (Austedo); Valbenazine
tetrabenazine (Xenazine ®); deutetrabenazine (Austedo ™); valbenazine (Ingrezza ™) EOCCO POLICY Po licy Type: PA/SP Pharmacy Coverage Policy: EOCCO157 Description Tetrabenazine (Xenazine), deutetrabenazine (Austedo) and valbenazine (Ingrezza) are reversible vesicular monoamine transporter 2 (VMAT2) inhibitors that act by regulating monoamine uptake from the cytoplasm to the synaptic vesicle. Its mechanism of action in Tardive dyskinesia or chorea-reduction is unknown . Length of Authorization Initial (Tardive dyskinesia): Three months Initial (Chorea associated with Huntington’s disease): 12 months Renewal: 12 months Quantity limits Product Name Dosage Form Indication Quantity Limit 12.5 mg Chorea associated with 60 tablets/30 days 25 mg Huntington’s disease Chorea associated with tetrabenazine (Xenazine) Huntington’s disease, 25 mg genotyped extensive 120 tablets/30 days and intermediate metabolizers 12.5 mg Chorea associated with 60 tablets/30 days 25 mg Huntington’s disease Chorea associated with generic tetrabenazine Huntington’s disease, 25 mg genotyped extensive 120 tablets/30 days and intermediate metabolizers 6 mg Tardive dyskinesia in adults; Chorea 30 tablets/30 days deutetrabenazine (Austedo) 9 mg associated with 12 mg Huntington’s disease 120 tablets/30 days 40 mg 30 capsules/30 days; valbenazine (Ingrezza) Tardive Dyskinesia 80 mg 4-week Initiation Pack Initial Evaluation I. Tetrabenazine (Xenazine), deutetrabenazine (Austedo) and valbenazine (Ingrezza) may be considered medically necessary when the following criteria below are met: A. Member is 18 years of age or older; AND B. Medication is prescribed by, or in consultation with, a neurologist or psychiatrist; AND C. Medication will not be used in combination with another VMAT2 inhibitor [e.g. tetrabenazine (Xenazine), deutetrabenazine (Austedo) valbenazine (Ingrezza)], monoamine oxidase inhibitor (MAOI) [e.g. -

Clinical Review, Adverse Events
Clinical Review, Adverse Events Drug: Carbamazepine NDA: 16-608, Tegretol 20-712, Carbatrol 21-710, Equetro Adverse Event: Stevens-Johnson Syndrome Reviewer: Ronald Farkas, MD, PhD Medical Reviewer, DNP, ODE I 1. Executive Summary 1.1 Background Carbamazepine (CBZ) is an anticonvulsant with FDA-approved indications in epilepsy, bipolar disorder and neuropathic pain. CBZ is associated with Stevens-Johnson syndrome (SJS) and Toxic Epidermal Necrolysis (TEN), closely related serious cutaneous adverse drug reactions that can be permanently disabling or fatal. Other anticonvulsants, including phenytoin, phenobarbital, and lamotrigine are also associated with SJS/TEN, as are members of a variety of other drug classes, including nonsteriodal anti-inflammatory drugs and sulfa drugs. The incidence of CBZ-associated SJS/TEN has been considered “extremely rare,” as noted in current U.S. drug labeling. However, recent publications and postmarketing data suggest that CBZ- associated SJS/TEN occurs at a much higher rate in some Asian populations, about 2.5 cases per 1,000 new exposures, and that most of this increased risk is in individuals carrying a specific human leukocyte antigen (HLA) allele, HLA-B*1502. This HLA-B allele is present in about 5- to 20% of many, but not all, Asian populations, and is also present in about 2- to 4% of South Asians/Indians. The allele is also present at a lower frequency, < 1%, in several other ethnic groups around the world (although likely due to distant Asian ancestry). About 10% of U.S. Asians carry HLA-B*1502. HLA-B*1502 is generally not present in the U.S. -

New Zealand Data Sheet 1
New Zealand Data Sheet 1. PRODUCT NAME Motetis 25 mg tablet 2. QUALITATIVE AND QUANTITATIVE COMPOSITION Each Motetis 25 mg tablets contains 25 mg of tetrabenazine. Excipient(s) with known effect Contains lactose monohydrate. For the full list of excipients, see Section 6.1. 3. PHARMACEUTICAL FORM Yellow, round, flat tablet with a score line on one side. The tablet can be divided into equal halves. 4. CLINICAL PARTICULARS 4.1. Therapeutic indications Movement disorders associated with organic central nervous system conditions, such as Huntington's chorea, hemiballismus and senile chorea. Tetrabenazine is also indicated for the treatment of moderate to severe tardive dyskinesia, which is disabling and/or socially embarrassing. The condition should be persistent despite withdrawal of antipsychotic therapy, or in cases where withdrawal of antipsychotic medication is not a realistic option; also, where the condition persists despite reduction in dosage of antipsychotic medication or switching to atypical antipsychotic medication. 4.2. Dose and method of administration Dose Proper dosing of tetrabenazine involves careful titration of therapy to determine an individualised dose for each patient. When first prescribed, tetrabenazine therapy should be titrated slowly over several weeks to allow the identification of a dose for chronic use that reduces chorea and is well tolerated. Movement disorders Dosage and administration are variable and only a guide is given. An initial starting dose of 25 mg three times a day is recommended. This can be increased by 25 mg a day every three (3) or four (4) days until 200 mg per day is being given or the limit of tolerance, as dictated by unwanted effects, is reached, whichever is the lower dose. -

Inhaled Loxapine Monograph
Inhaled Loxapine Monograph Inhaled Loxapine (ADASUVE) National Drug Monograph February 2015 VA Pharmacy Benefits Management Services, Medical Advisory Panel, and VISN Pharmacist Executives The purpose of VA PBM Services drug monographs is to provide a comprehensive drug review for making formulary decisions. Updates will be made when new clinical data warrant additional formulary discussion. Documents will be placed in the Archive section when the information is deemed to be no longer current. FDA Approval Information Description/Mechanism of Inhaled loxapine is a typical antipsychotic used in the treatment of acute Action agitation associated with schizophrenia and bipolar I disorder in adults. Loxapine’s mechanism of action for reducing agitation in schizophrenia and bipolar I disorder is unknown. Its effects are thought to be mediated through blocking postsynaptic dopamine D2 receptors as well as some activity at the serotonin 5-HT2A receptors. Indication(s) Under Review in Inhaled loxapine is a typical antipsychotic indicated for the acute treatment of this document (may include agitation associated with schizophrenia or bipolar I disorder in adults. off label) Off-label use Agitation related to any other cause not due to schizophrenia and bipolar I disorder. Dosage Form(s) Under 10mg oral inhalation using a new STACCATO inhaler device. Review REMS REMS No REMS Post-marketing Study Required See Other Considerations for additional REMS information Pregnancy Rating C Executive Summary Efficacy Inhaled loxapine was superior to placebo in reducing acute agitation at 2 hours post dose measured by the Positive and Negative Syndrome Scale-Excited Component (PEC) in patients with bipolar I disorder and schizophrenia. -

Mrx CLINICAL ALERT
MRx AUGUST 2018 CLINICAL ALERT YOUR MONTHLY SOURCE FOR DRUG INFORMATION HIGHLIGHTS HOT TOPIC: potential to placebo in non-dependent FIRST DRUG COMPRISED adult recreational drug users. EDITORIAL OF ACTIVE MARIJUANA DERIVATIVE APPROVED Cannabidiol oral solution was studied STAFF in 3 randomized, double-blind, placebo- Under Priority review, the United States controlled clinical trials including 516 (US) Food and Drug Administration (FDA) patients with either LGS or Dravet EDITOR IN CHIEF approved cannabidiol oral solution syndrome. Cannabidiol taken with ® Maryam Tabatabai (Epidiolex ) for the treatment of the patient’s current anticonvulsant PharmD seizures associated with Lennox-Gastaut regimen was shown to be effective syndrome (LGS) or Dravet syndrome in in reducing the frequency of seizures patients ≥ 2 years of age. It is the only when compared with placebo. Over EXECUTIVE EDITOR drug approved to treat Dravet syndrome the 12-week treatment period, the Carole Kerzic and the FDA designated it as an Orphan reduction in median number of LGS RPh drug and Rare pediatric disease drug seizures ranged from 37% to 44% with to treat both serious, difficult-to-treat CBD and 17% to 22% with placebo. DEPUTY EDITORS childhood seizure disorders. Dravet For Dravet syndrome, seizure reductions Stephanie Christofferson syndrome is diagnosed in approximately were 39% and 13% with CBD and PharmD 1 in 15,700 individuals in the US and placebo, respectively. Common side is characterized by frequent prolonged effects (≥ 10%) reported with CBD Jessica Czechowski seizures and developmental delays. LGS were increased liver transaminases PharmD involves frequent drop seizures and (particularly with concurrent use of impaired intellectual development.