Inside This Issue
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

No. 33981 2 No
Pretoria, 4 February 2011 Februarle No. 33981 2 No. 33981 GOVERNMENT GAZETTE, 4 FEBRUARY 2011 IMPORTANT NOTICE The Government Printing Works will not be held responsible for faxed documents not received due to errors on the fax machine or faxes received which are unclear or incomplete. Please be advised that an "OK" slip, received from a fax machine, will not be accepted as proof that documents were received by the GPW for printing. If documents are faxed to the GPW it will be the sender's respon sibility to phone and confirm that the documents were received in good order. Furthermore the Government Printing Works will also not be held responsible for cancellations and amendments which have not been done on original documents received from clients. CONTENTS INHOUD Bladsy Koerant Page Gazette No. No. No. No. No. No. GENERAL NOTICE ALGEMENEKENNISGEWING Health, Department of Gesondheld, Departement van General Notice A/gemene Kennisgewing 58 Medicines and Related Substances Act 58 Wet op Beheer van Medisyne en (101/1965): Medicines Control Council: Verwante Stowwe (101/1965): Conditions of registration of a medicine Medisynebeheerraad: Voorwaardes vir in terms of the provisions of section die registrasie van 'n medisyne in terme 15 (7) ..................................................... .. 3 33981 van die bepalings van artikel 15 (7) ........ 4 33981 STAATSKOERANT, 4 FEBRUARIE 2011 No. 33981 3 GENERAL NOTICE ALGEMENE KENNISGEWING NOTICE 58 OF 2011 MEDICINES CONTROL COUNCIL CONDITIONS OF REGISTRATION OF A MEDICINE IN TERMS OF THE PROVISIONS OF SECTION 15(7) OF THE MEDICINES AND RELATED SUBSTANCES ACT, 1965 (ACT 101 OF 1965) 1. The applicant shall ensure that the medicine is manufactured and controlled in terms of the current Good Manufacturing Practices as determined by Council 2. -

Mcneil Consumer : Mdl No
IN THE UNITED STATES DISTRICT COURT FOR THE EASTERN DISTRICT OF PENNSYLVANIA IN RE: MCNEIL CONSUMER : MDL NO. 2190 HEALTHCARE, ET AL., MARKETING : AND SALES PRACTICES LITIGATION : : Applies to: : ALL ACTIONS : MEMORANDUM McLaughlin, J. July 13, 2012 This multidistrict litigation arises out of quality control problems at the defendants’ facility manufacturing over- the-counter healthcare products in Fort Washington, Pennsylvania, which led to a series of recalls of those products. The named plaintiffs assert claims for economic loss on behalf of a putative nationwide class against Johnson & Johnson (“J&J”), McNeil Consumer Healthcare (“McNeil”), and four of their executives. The plaintiffs allege that they overpaid for the defendants’ products as a result of the recalls and the defendants’ scheme to conceal or downplay the scope of the quality control problems. The defendants, who have offered a coupon or cash refund to consumers who purchased recalled drugs, have moved to dismiss the operative complaint, and assert that the named plaintiffs lack constitutional standing and have not met the applicable pleading standard. The Court will grant the defendants’ motion because the plaintiffs have not pled facts that show a cognizable injury in fact, which is required to confer Article III standing. I. Procedural Background This litigation resulted from the consolidation of ten individual actions filed around the country. Haviland v. McNeil Consumer Healthcare, No. 10-2195, was filed in this Court on May 12, 2010, asserting economic injuries arising out of the April 30, 2010 recall of over-the-counter children’s drugs by McNeil, a part of the J&J “Family of Companies.” Eight additional cases, also arising out of the April 2010 recall, were filed in district courts around the country.1 All cases asserted claims for economic injury only, with the exception of Rivera v. -

9894 Pharma Tech Media Planner V6 2007
www.pharmtech.com years 1977– 2007 30ANNIVERSARY CELEBRATING 30 YEARS AS THE 2007 INDUSTRY’S MOST AUTHORITATIVE SOURCE Media Planner years 1977–2007 ANNIVERSARY years 1977–2007 ANNIVERSARY THE PHARMACEUTICAL TECHNOLOGY BRAND PUBLISHER’S STATEMENT Pharmaceutical Technology’s authoritative reputation and powerful brand recognition within the pharmaceutical/biopharmaceutical development & manufacturing marketplace will help you establish and maintain your own strong brand among pharma industry decision makers. A circulation of 38,667 BPA-qualified subscribers* and unmatched peer written and reviewed editorial make Pharmaceutical Technology an invaluable resource within top pharma companies, as well as small, specialty and biotech pharma companies spending billions each year on pharmaceutical development and manufacturing. Please celebrate with us as Pharmaceutical Technology marks its 30th Anniversary as the industry leader. —Michael Tracey, Publisher % 90 of readers rated Pharmaceutical Technology as important or very important to them as a professionalˆ EDITORIAL MISSION Pharmaceutical Technology publishes authoritative, reliable, and timely peer-reviewed research and expert analyses for scientists, engineers, technicians, and managers engaged in process development, manufacturing, formulation, analytical technology, packaging and regulatory compliance in the pharmaceutical and biotechnology industries. —Douglas McCormick, Editor in chief www.pharmtech.com *BPA June 2006 Statement ^2006 Readership Study Conducted by Advanstar Research -

Notice Under S66 of the Commerce Act 1986 Application by Johnson & Johnson to Acquire the Stock, Assets and Business Of
PUBLIC COPY Notice under s66 of the Commerce Act 1986 Application by Johnson & Johnson to acquire the stock, assets and business of the Consumer Healthcare division of Pfizer Inc. COMMERCE ACT 1986: BUSINESS ACQUISITION SECTION 66: NOTICE SEEKING CLEARANCE 28 September 2006 The Registrar Business Acquisitions and Authorisations Commerce Commission PO Box 2351 Wellington Pursuant to s66(1) of the Commerce Act 1986 notice is hereby given seeking clearance of a proposed business acquisition. 518689_1.DOC 2 CONTENTS EXECUTIVE SUMMARY PART 1: TRANSACTION DETAILS 1 The business acquisition for which clearance is sought 2 The person giving this notice 3 Confidentiality 4 Participants 5 Interconnected and associated persons 6 Beneficial interests 7 Links between participants 8 Common directorships 9 Business activities of the participants 10 Reasons for the proposed acquisition PART II: IDENTIFICATION OF MARKETS AFFECTED 11 Horizontal aggregation 12 Differentiated product markets 13 Differentiated product markets 14 Vertical integration 15 Other business acquisitions PARTS III, IV AND V: CONSTRAINTS ON MARKET POWERS BY EXISTING AND POTENTIAL COMPETITION AND OTHER POTENTIAL CONSTRAINTS 16 Allergy medication 17 Products for the treatment of worms 18 Thrush treatment CERTIFICATE APPENDICES 1. Heartburn and indigestion remedies (MYLANTA and MOTILIUM) 2. Worm treatments (COMBANTRIN, VERMOX) 3. Cold, flu, nasal decongestant, cough relief and sort throat medications (CODRAL, SINUTAB, SUDAFED, BENADRYL, BRONDECON) 4. Allergy relief products (ACTIFED, SINUTAB, SUDAFED, VISINE, LIVOSTIN) 518689_1.DOC 3 5. Thrush treatments (DIFLUCAN ONE, DAKTARIN, DAKTAGOLD, NIZORAL, SPORANOX) 6. Shampoo (PREGAINE, ROGAINE, NEUTROGENA, JOHNSON’S BABY SHAMPOO) 7. Hand hygiene (PURELL and MICRO SHIELD) 8. Competitor worm treatment products 9. Multinational pharmaceutical businesses: GlaxoSmithKline, Douglas Pharmaceuticals, Alphapharm, Bayer Group 10. -

Medication Code Key: PMCODE and Drug Name in 2007 NHHCS Cdc-Pdf
Medication Code Key: PMCODE and Drug Name in 2007 NHHCS PMCODE Drug Name 00002 TAMIFLU 00003 DITROPAN XL II 00004 LIDODERM PATCH 00008 VIACTIV 00010 A AND D II 00013 MYCOPHENOLATE MOFETIL 00017 SIROLIMUS 00019 HAWTHORN 00027 SYNAGIS 00032 EXCEDRIN MIGRAINE 00036 MAALOX PLUS 00037 ACEON 00038 GLYSET 00039 SONATA 00042 PROTONIX 00044 PANLOR DC 00048 MOBIC 00052 SILDENAFIL CITRATE 00053 TAMSULOSIN HYDROCHLORIDE 00054 COMTAN 00058 MINERAL SUPPLEMENT 00061 BISMUTH 00071 CERTAVITE 00073 LUXIQ 00075 SAL-TROPINE 00076 TRILEPTAL 00078 AGGRENOX 00080 CARBIDOPA-LEVODOPA 00081 EXELON 00084 PREGABALIN 00085 ORAMORPH 00096 OSTEO-BIFLEX 00099 ALOCRIL 00100 A.S.A. 00101 ISOSORBIDE DINITRATE 00102 ISOSORBIDE MONONITRATE 00107 ROSIGLITAZONE MALEATE 00109 URSODIOL 00112 MEDERMA 00113 ANDROGEL 00114 DILTIA XT 00117 CRANBERRY 00123 NICOTINE 00125 AVELOX 00132 CAL-MAG 00133 CANDESARTAN Page 1 Medication Code Key: PMCODE and Drug Name in 2007 NHHCS PMCODE Drug Name 00148 PROLIXIN D 00149 D51/2 NS 00150 NICODERM CQ PATCH 00151 TUSSIN 00152 CEREZYME 00154 CHILDREN'S IBUPROFEN 00156 PROPOXACET-N 00159 KALETRA 00161 BISOPROLOL 00167 NOVOLIN N 00169 KETOROLAC TROMETHAMINE 00172 OPHTHALMIC OINTMENT 00173 ELA-MAX 00176 PREDNISOLONE ACETATE 00179 COLLOID SILVER 00184 KEPPRA 00187 OPHTHALMIC DROPS 00190 ABDEC 00191 HAPONAL 00192 SPECTRAVITE 00198 ENOXAPARIN SODIUM 00206 ACTONEL 00208 CELECOXIB 00209 GLUCOVANCE 00211 LEVALL 5.0 00213 PANTOPRAZOLE SODIUM 00217 TEMODAR 00218 CARBAMIDE PEROXIDE 00221 CHINESE HERBAL MEDS 00224 MILK AND MOLASSES ENEMA 00238 ZOLMITRIPTAN 00239 -
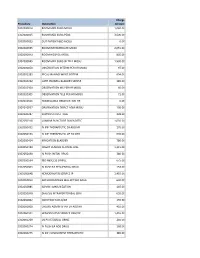
Procedure Description Charge Amount 0302000014 ROOM MED
Charge Procedure Description Amount 0302000014 ROOM MED SURG MOSU 1,040.00 0302000015 ROOM MED SURG PEDS 3,500.00 0302000033 OUT PATIENT BED MOSU 0.00 0302000035 ROOM INTERMEDIATE MOSU 2,055.00 0302000043 ROOM HOSPICE MOSU 805.00 0302000045 ROOM MED SURG W TELE MOSU 1,530.00 0302000050 OBSERVATION INTERM PER HR MOSU 97.00 0302002283 FECAL MANAGEMENT SYSTEM 694.00 0302010232 CATH INDWELL BLADDER SIMPLE 185.00 0302010304 OBSERVATION MS PER HR MOSU 60.00 0302010305 OBSERVATION TELE PER HR MOSU 71.00 0302010556 NONBILLABLE OBSERVATION HR 0.00 0302010557 OBSERVATION DIRECT ADM MOSU 130.00 0302020287 SUPPLIES CHEST TUBE 228.00 0302050318 LUMBAR PUNCTURE DIAGNOSTIC 1,035.00 0302050451 IV INF THERAPEUTIC EA ADD HR 170.00 0302050453 IV INF THERAPEUTIC UP TO 1HR 635.00 0302050454 IRRIGATION BLADDER 780.00 0302050480 INSERT VENOUS CENTRAL LINE 1,315.00 0302050490 IV PUSH INITIAL DRUG 380.00 0302050534 I&D ABSCESS SIMPLE 675.00 0302050603 IV PUSH EA SEQUENTIAL DRUG 153.00 0302050648 HEMODIALYSIS SERVICE IP 1,455.00 0302050813 ARTHROCENTESIS MAJ JNT WO IMAG 630.00 0302050885 ADMIN IMMUNIZATION 145.00 0302050948 DIALYSIS INTRAPERITONEAL SERV 655.00 0302060002 INJECTION SUB-Q/IM 155.00 0302060008 CHEMO ADMIN IV INF EA ADD HR 410.00 0302060101 HEMODIALYSIS SERVICE OBS/OP 1,455.00 0302060269 US PV RESIDUAL URINE 240.00 0302060274 IV PUSH EA ADD DRUG 168.00 0302060275 IV INF CONCURRENT THERAPEUTIC 385.00 0302060276 IV INF SEQUENTIAL THER UP TO 1 191.00 0302060293 INSERT STRAIGHT CATH THERAPEUT 185.00 0302060372 CHEMO ADMIN IV INF SEQ 1 HR 525.00 0302060373 -

TITLE Safety and Efficacy of Over-The-Counter Drug Use by the Elderly
" DOCUMENT RESUME ED 245 148 CG 017 518 TITLE Safety and Efficacy of Over-the-Counter Drug Use by the Elderly. Hearing before the Subcommittee on Health and Long-Term Care of the Select Committee on Aging. House of Representatives, Ninety-Eighth Congress, First Session. INSTITUTION Congress of the U.S., Washington, D.C. House Select Committee on Aging. REPORT NO House-Comm-Pub-98-409 PUB DATE 21 Jul 83 NOTE 507p.; Some pages are marginally legible due to small print. PUB TYPE Legal/Legislative/Regulatory Materials (090) EDRS PRICE MF02 Plus Postage. PC Not Available from EDRS. DESCRIPTORS *Consumer Protection; Dietetics; Drug Legislation; *Drug Use; Gerontology; Hearings; *Older Adults; Physical Health; *Safety IDENTIFIERS Congress 98th; Long Term Care; *Nonprescription Drugs; *PPA ABSTRACT This document contains the prepared statements and panel testimony from the Congressional hearing on over-the-counter (OTC) drug use by the elderly. Opening statements are given by Representatives Claude Pepper (chairman), Ralph Regula, Mary Rose Oakar, Michael Bilirakis, Tom Lantos, and Hal Daub. Topics which are covered include the incidence and quantity of drug use by the elderly, health risks, adverse reactions, phenylpropanolamine (PPA), consumer protection, and the Federal Drug Administration's (FDA) role in the OTC drug safety and reguIatiem. Testimony of the first panel on OTC drugs, particularly weight reduction medications containing PPA, is given by representatives of the Health Research Group, the National Broadcasting Company, and consumers. Testimony of the second panel on mail fraud schemes perpetrated against senior citizens is given by consumer advocates representing the United States Postal Service, Criminal Investigations and Consumer Protection Divisions, and the Center for Science in the Public Interest. -

Memorandum JUL 1 6 201D
Memorandum Subject Date Additional Quota Letters Received by July 15, 2010 (DFN: 630-08.2) JUL 1 6 201D To From Christine A. Sanncrud. Ph.D., Chief "Barbar• J.Illoockholdt, Chief Drug & Chemical Evaluation Section Regul• torn Section Office of Diverison Control Off cL rl Diversion Control On July 15. 2010, this section received your e-mail requesting a review of seventeen (17) quota applications from sixteen (16) registered manufacturers to determine if there arc any pending administrative/legal actions against these applicants and to advise ODE of the findings. ODOR conducted reviews (NADDIS, CSA, etc), as well as surveyed the responsible field offices for their input and recommendations. Provided below are the results and recommendations. QUOTA APPLICANTS wan NO ADVERSE OR DEROGATORY INFORMATIO N Novartis Consumer I lealth Lincoln (10345) (b)(4);(b)(7)(E) Baxter (10346) Generics Bidco li bda Vintage (10347) Noramco Delaware (10348) Pharmaceuticals International inc. (10349) Pharmedium (10351) Pharmedium (10352) Rhodes (10356) Bio-Pharm (10357) Patheon (10358) Patheon (10359) Watson (10361) B & B (10363) lospira. Inc. NC (10364) Epic Pharma (10366) Mallinckrodt I lobart (10367) Chemtos (10368) Vol. II Page 55 2 Per consultation with the field offices. DEA does not have sufficient grounds to limit, restrict. or deny quota requests from these registrants. Based on this information. ODG suggests that you proceed with the completion of the quota applications. If you have any questions pertaining to this information. please feel free to contact me (b)(6);(b)(7)( or SC C) (b)(6);(b)(7)(C) Vol. II Page 56 Memorandum Subject Date Additional Quota Letters Received as of July 19, 201() (DFN: 630-08.2) stir 2 8 2010,., To Fr ,./"./ Christine A. -

Otc) Benefit Medication Formulary
OVER-THE-COUNTER (OTC) BENEFIT MEDICATION FORMULARY The OTC formulary is a list of medicines (Schedule 0-3) that will be paid from the OTC benefit, subject to the limit specified by the scheme. Metropolitan Generic Reference Price (METREF) will apply. Co-payments will apply for items amounting to more than the reference price. The formulary is subject to regular review. Metropolitan Health reserves the right to update and change the formulary when new information becomes available. IMPORTANT: Please note that the medication on this list is published on an annual basis. Medication and prices are subject to change based on new clinical information and/or pricing updates which may not be reflected on the list below. Product name Active Ingredient Strength Dosage form Schedule Nappi code Oral antiseptics and sore throat Andolex C Benzydamide SPR 1 710149 Andolex Oral Rinse Benzydamide . MOW 1 810576 Andolex Spray Benzydamide . SPR 1 816167 Betadine oral Povidone-iodine MOW 0 707937 Dermadine oral garg Povidone-iodine 1g/100ml MOW 0 799688 Iodised throat Antiseptics LOZ 1 892670 Iodised throat Antiseptics LOZ 1 810010 Medi-keel A Cetylpyridinium LOZ 1 741035 Medi-Keel A black currant Cetylpyridinium LOZ 1 884233 Medi-Keel A honey & lemon Cetylpyridinium LOZ 1 884225 Podine mouth wash Povidone-iodine MOW 0 785210 Septadine oral antiseptic Povidone-iodine MOW 0 819859 Steridine mouthwash and gargle liquid Povidone-iodine MOW 0 890365 Tcp antiseptic Antiseptics SLN 1 815950 Throflam Benzydamide MOW 1 712171 Throflam Benzydamine 22.5mg/15ml -

Pharmaceutical Research and Manufacturers of America – Phrma
The Short-Term and Long-Term Competitive Impact of Authorized Generics A Report for the Federal Trade Commission October 28, 2009 TABLE OF CONTENTS INTRODUCTION ........................................................................................................................... 1 DISCUSSION .................................................................................................................................. 3 I. LONG-TERM COMPETITIVE HARM IS UNLIKELY ................................................... 3 A. Unsupported Foreclosure Claims Have Been Made For Nearly Two Decades .................................................................................................................... 4 B. Marketplace Realities Undercut The Long-Tenn Foreclosure Theory .................... 7 1. Sales and Profitability Are Growing ........................................................... 7 2. Wall Street Valuations Are Growing ........................................................ 10 II. THE FTC PRICING ANALYSIS SHOWS $880 MILLION IN CONSUMER SAVINGS .......................................................................................................................... 14 III. CONSUMER SAVINGS SHOULD BE MEASURED USING WHOLESALE DATA AND WEIGHTED AVERAGE PRICES .............................................................. 17 A. The Wholesale Data Carries Far More Weight.. .................................................... 17 B. Average Drug Prices Should Be Volume-Weighted ............................................. 19 IV. -

Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Patent Drugs Under Patent Drugs Under Paten
Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under P Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under P Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under P Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patents Drugs Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under P Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Patent DrugsDrug Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Under Patent Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under P Drugs Under Patent Drugs Under Patent Drugs Under Patent Drugs Under -

Clinical Research Services
Clinical Research Services Advancing the future of medicine through research. Who We Are Experience Clinical Research Services is a department within Clinical Research Services has been involved CHI St. Alexius Health in Bismarck, ND. We are fully in clinical research since 1987. Our professional dedicated to providing research support services staff has more than 180 years of combined to physicians within our region and the valued experience in clinical trials. We are committed patients they serve. to providing outstanding service to ensure the success of every project. CHI St. Alexius Health is a tertiary care facility affiliated with PrimeCare, a health group which Our involvement in inpatient and outpatient combines the most experienced, trusted, and phase II-IV clinical and device research studies has proven medical leaders in the area. PrimeCare is contributed substantially to the approval of new comprised of a network which includes more than drugs and treatments. You should carefully consider 190 physicians in private and institutional practice, both the benefits and the risks of participation including: CHI St. Alexius Health, Mid Dakota Clinic, before enrolling in a study. The Bone & Joint Center, and other affiliated area physicians. Many of these physicians are actively Previous clinical study trials: involved as investigators for clinical trials. • Oncology • Orthopaedic • Rheumatology • Diabetes Our clinical research team is committed to providing: • Neurology • Weight loss • Rapid study start-up • Cardiology • Pain • Pro-active patient enrollment • Urology • Men’s Health • Clean data submission • Gastroenterology • Women’s Health • Highly experienced principal • Infectious Disease • Medical Devices investigators • Pediatric • Initial training and continuing education for all support personnel Sponsors Patient Demographics Abbott Laboratories Characteristics of our patients include Acorda Therapeutics, Inc.