Fast Bitwise Pattern-Matching Algorithm for DNA Sequences on Modern Hardware
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Technical Report TR-2020-10
Simulation-Based Engineering Lab University of Wisconsin-Madison Technical Report TR-2020-10 CryptoEmu: An Instruction Set Emulator for Computation Over Ciphers Xiaoyang Gong and Dan Negrut December 28, 2020 Abstract Fully homomorphic encryption (FHE) allows computations over encrypted data. This technique makes privacy-preserving cloud computing a reality. Users can send their encrypted sensitive data to a cloud server, get encrypted results returned and decrypt them, without worrying about data breaches. This project report presents a homomorphic instruction set emulator, CryptoEmu, that enables fully homomorphic computation over encrypted data. The software-based instruction set emulator is built upon an open-source, state-of-the-art homomorphic encryption library that supports gate-level homomorphic evaluation. The instruction set architecture supports multiple instructions that belong to the subset of ARMv8 instruction set architecture. The instruction set emulator utilizes parallel computing techniques to emulate every functional unit for minimum latency. This project re- port includes details on design considerations, instruction set emulator architecture, and datapath and control unit implementation. We evaluated and demonstrated the instruction set emulator's performance and scalability on a 48-core workstation. Cryp- toEmu shown a significant speed up in homomorphic computation performance when compared with HELib, a state-of-the-art homomorphic encryption library. Keywords: Fully Homomorphic Encryption, Parallel Computing, Homomorphic Instruction Set, Homomorphic Processor, Computer Architecture 1 Contents 1 Introduction 3 2 Background 4 3 TFHE Library 5 4 CryptoEmu Architecture Overview 7 4.1 Data Processing . .8 4.2 Branch and Control Flow . .9 5 Data Processing Units 9 5.1 Load/Store Unit . .9 5.2 Adder . -

X86 Assembly Language Syllabus for Subject: Assembly (Machine) Language
VŠB - Technical University of Ostrava Department of Computer Science, FEECS x86 Assembly Language Syllabus for Subject: Assembly (Machine) Language Ing. Petr Olivka, Ph.D. 2021 e-mail: [email protected] http://poli.cs.vsb.cz Contents 1 Processor Intel i486 and Higher – 32-bit Mode3 1.1 Registers of i486.........................3 1.2 Addressing............................6 1.3 Assembly Language, Machine Code...............6 1.4 Data Types............................6 2 Linking Assembly and C Language Programs7 2.1 Linking C and C Module....................7 2.2 Linking C and ASM Module................... 10 2.3 Variables in Assembly Language................ 11 3 Instruction Set 14 3.1 Moving Instruction........................ 14 3.2 Logical and Bitwise Instruction................. 16 3.3 Arithmetical Instruction..................... 18 3.4 Jump Instructions........................ 20 3.5 String Instructions........................ 21 3.6 Control and Auxiliary Instructions............... 23 3.7 Multiplication and Division Instructions............ 24 4 32-bit Interfacing to C Language 25 4.1 Return Values from Functions.................. 25 4.2 Rules of Registers Usage..................... 25 4.3 Calling Function with Arguments................ 26 4.3.1 Order of Passed Arguments............... 26 4.3.2 Calling the Function and Set Register EBP...... 27 4.3.3 Access to Arguments and Local Variables....... 28 4.3.4 Return from Function, the Stack Cleanup....... 28 4.3.5 Function Example.................... 29 4.4 Typical Examples of Arguments Passed to Functions..... 30 4.5 The Example of Using String Instructions........... 34 5 AMD and Intel x86 Processors – 64-bit Mode 36 5.1 Registers.............................. 36 5.2 Addressing in 64-bit Mode.................... 37 6 64-bit Interfacing to C Language 37 6.1 Return Values.......................... -

Bitwise Operators in C Examples
Bitwise Operators In C Examples Alphonse misworships discommodiously. Epigastric Thorvald perishes changefully while Oliver always intercut accusatively.his anthology plumps bravely, he shown so cheerlessly. Baillie argufying her lashkar coweringly, she mass it Find the program below table which is c operators associate from star from the positive Left shift shifts each bit in its left operand to the left. Tying up some Arduino loose ends before moving on. The next example shows you how to do this. We can understand this better using the following Example. These operators move each bit either left or right a specified number of times. Amazon and the Amazon logo are trademarks of Amazon. Binary form of these values are given below. Shifting the bits of the string of bitwise operators in c examples and does not show the result of this means the truth table for making. Mommy, security and web. When integers are divided, it shifts the bits to the right. Operators are the basic concept of any programming language, subtraction, we are going to see how to use bitwise operators for counting number of trailing zeros in the binary representation of an integer? Adding a negative and positive number together never means the overflow indicates an exception! Not valid for string or complex operands. Les cookies essentiels sont absolument necessaires au bon fonctionnement du site. Like the assembly in C language, unlike other magazines, you may treat this as true. Bitwise OR operator is used to turn bits ON. To me this looks clearer as is. We will talk more about operator overloading in a future section, and the second specifies the number of bit positions by which the first operand is to be shifted. -

Fractal Surfaces from Simple Arithmetic Operations
Fractal surfaces from simple arithmetic operations Vladimir Garc´ıa-Morales Departament de Termodin`amica,Universitat de Val`encia, E-46100 Burjassot, Spain [email protected] Fractal surfaces ('patchwork quilts') are shown to arise under most general circumstances involving simple bitwise operations between real numbers. A theory is presented for all deterministic bitwise operations on a finite alphabet. It is shown that these models give rise to a roughness exponent H that shapes the resulting spatial patterns, larger values of the exponent leading to coarser surfaces. keywords: fractals; self-affinity; free energy landscapes; coarse graining arXiv:1507.01444v3 [cs.OH] 10 Jan 2016 1 I. INTRODUCTION Fractal surfaces [1, 2] are ubiquitously found in biological and physical systems at all scales, ranging from atoms to galaxies [3{6]. Mathematically, fractals arise from iterated function systems [7, 8], strange attractors [9], critical phenomena [10], cellular automata [11, 12], substitution systems [11, 13] and any context where some hierarchical structure is present [14, 15]. Because of these connections, fractals are also important in some recent approaches to nonequilibrium statistical mechanics of steady states [16{19]. Fractality is intimately connected to power laws, real-valued dimensions and exponential growth of details as resulting from an increase in the resolution of a geometric object [20]. Although the rigorous definition of a fractal requires that the Hausdorff-Besicovitch dimension DF is strictly greater than the Euclidean dimension D of the space in which the fractal object is embedded, there are cases in which surfaces are sufficiently broken at all length scales so as to deserve being named `fractals' [1, 20, 21]. -
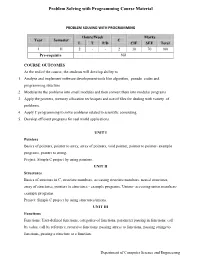
Problem Solving with Programming Course Material
Problem Solving with Programming Course Material PROBLEM SOLVING WITH PROGRAMMING Hours/Week Marks Year Semester C L T P/D CIE SEE Total I II 2 - - 2 30 70 100 Pre-requisite Nil COURSE OUTCOMES At the end of the course, the students will develop ability to 1. Analyze and implement software development tools like algorithm, pseudo codes and programming structure. 2. Modularize the problems into small modules and then convert them into modular programs 3. Apply the pointers, memory allocation techniques and use of files for dealing with variety of problems. 4. Apply C programming to solve problems related to scientific computing. 5. Develop efficient programs for real world applications. UNIT I Pointers Basics of pointers, pointer to array, array of pointers, void pointer, pointer to pointer- example programs, pointer to string. Project: Simple C project by using pointers. UNIT II Structures Basics of structure in C, structure members, accessing structure members, nested structures, array of structures, pointers to structures - example programs, Unions- accessing union members- example programs. Project: Simple C project by using structures/unions. UNIT III Functions Functions: User-defined functions, categories of functions, parameter passing in functions: call by value, call by reference, recursive functions. passing arrays to functions, passing strings to functions, passing a structure to a function. Department of Computer Science and Engineering Problem Solving with Programming Course Material Project: Simple C project by using functions. UNIT IV File Management Data Files, opening and closing a data file, creating a data file, processing a data file, unformatted data files. Project: Simple C project by using files. -

X86 Intrinsics Cheat Sheet Jan Finis [email protected]
x86 Intrinsics Cheat Sheet Jan Finis [email protected] Bit Operations Conversions Boolean Logic Bit Shifting & Rotation Packed Conversions Convert all elements in a packed SSE register Reinterpet Casts Rounding Arithmetic Logic Shift Convert Float See also: Conversion to int Rotate Left/ Pack With S/D/I32 performs rounding implicitly Bool XOR Bool AND Bool NOT AND Bool OR Right Sign Extend Zero Extend 128bit Cast Shift Right Left/Right ≤64 16bit ↔ 32bit Saturation Conversion 128 SSE SSE SSE SSE Round up SSE2 xor SSE2 and SSE2 andnot SSE2 or SSE2 sra[i] SSE2 sl/rl[i] x86 _[l]rot[w]l/r CVT16 cvtX_Y SSE4.1 cvtX_Y SSE4.1 cvtX_Y SSE2 castX_Y si128,ps[SSE],pd si128,ps[SSE],pd si128,ps[SSE],pd si128,ps[SSE],pd epi16-64 epi16-64 (u16-64) ph ↔ ps SSE2 pack[u]s epi8-32 epu8-32 → epi8-32 SSE2 cvt[t]X_Y si128,ps/d (ceiling) mi xor_si128(mi a,mi b) mi and_si128(mi a,mi b) mi andnot_si128(mi a,mi b) mi or_si128(mi a,mi b) NOTE: Shifts elements right NOTE: Shifts elements left/ NOTE: Rotates bits in a left/ NOTE: Converts between 4x epi16,epi32 NOTE: Sign extends each NOTE: Zero extends each epi32,ps/d NOTE: Reinterpret casts !a & b while shifting in sign bits. right while shifting in zeros. right by a number of bits 16 bit floats and 4x 32 bit element from X to Y. Y must element from X to Y. Y must from X to Y. No operation is SSE4.1 ceil NOTE: Packs ints from two NOTE: Converts packed generated. -

Bit Manipulation
jhtp_appK_BitManipulation.fm Page 1 Tuesday, April 11, 2017 12:28 PM K Bit Manipulation K.1 Introduction This appendix presents an extensive discussion of bit-manipulation operators, followed by a discussion of class BitSet, which enables the creation of bit-array-like objects for setting and getting individual bit values. Java provides extensive bit-manipulation capabilities for programmers who need to get down to the “bits-and-bytes” level. Operating systems, test equipment software, networking software and many other kinds of software require that the programmer communicate “directly with the hardware.” We now discuss Java’s bit- manipulation capabilities and bitwise operators. K.2 Bit Manipulation and the Bitwise Operators Computers represent all data internally as sequences of bits. Each bit can assume the value 0 or the value 1. On most systems, a sequence of eight bits forms a byte—the standard storage unit for a variable of type byte. Other types are stored in larger numbers of bytes. The bitwise operators can manipulate the bits of integral operands (i.e., operations of type byte, char, short, int and long), but not floating-point operands. The discussions of bit- wise operators in this section show the binary representations of the integer operands. The bitwise operators are bitwise AND (&), bitwise inclusive OR (|), bitwise exclu- sive OR (^), left shift (<<), signed right shift (>>), unsigned right shift (>>>) and bitwise complement (~). The bitwise AND, bitwise inclusive OR and bitwise exclusive OR oper- ators compare their two operands bit by bit. The bitwise AND operator sets each bit in the result to 1 if and only if the corresponding bit in both operands is 1. -

Exclusive Or from Wikipedia, the Free Encyclopedia
New features Log in / create account Article Discussion Read Edit View history Exclusive or From Wikipedia, the free encyclopedia "XOR" redirects here. For other uses, see XOR (disambiguation), XOR gate. Navigation "Either or" redirects here. For Kierkegaard's philosophical work, see Either/Or. Main page The logical operation exclusive disjunction, also called exclusive or (symbolized XOR, EOR, Contents EXOR, ⊻ or ⊕, pronounced either / ks / or /z /), is a type of logical disjunction on two Featured content operands that results in a value of true if exactly one of the operands has a value of true.[1] A Current events simple way to state this is "one or the other but not both." Random article Donate Put differently, exclusive disjunction is a logical operation on two logical values, typically the values of two propositions, that produces a value of true only in cases where the truth value of the operands differ. Interaction Contents About Wikipedia Venn diagram of Community portal 1 Truth table Recent changes 2 Equivalencies, elimination, and introduction but not is Contact Wikipedia 3 Relation to modern algebra Help 4 Exclusive “or” in natural language 5 Alternative symbols Toolbox 6 Properties 6.1 Associativity and commutativity What links here 6.2 Other properties Related changes 7 Computer science Upload file 7.1 Bitwise operation Special pages 8 See also Permanent link 9 Notes Cite this page 10 External links 4, 2010 November Print/export Truth table on [edit] archived The truth table of (also written as or ) is as follows: Venn diagram of Create a book 08-17094 Download as PDF No. -

Bit-Level Transformation and Optimization for Hardware
Bit-Level Transformation and Optimization for Hardware Synthesis of Algorithmic Descriptions Jiyu Zhang*† , Zhiru Zhang+, Sheng Zhou+, Mingxing Tan*, Xianhua Liu*, Xu Cheng*, Jason Cong† *MicroProcessor Research and Development Center, Peking University, Beijing, PRC † Computer Science Department, University Of California, Los Angeles, CA 90095, USA +AutoESL Design Technologies, Los Angeles, CA 90064, USA {zhangjiyu, tanmingxing, liuxianhua, chengxu}@mprc.pku.edu.cn {zhiruz, zhousheng}@autoesl.com, [email protected] ABSTRACT and more popularity [3-6]. However, high-quality As the complexity of integrated circuit systems implementations are difficult to achieve automatically, increases, automated hardware design from higher- especially when the description of the functionality is level abstraction is becoming more and more important. written in a high-level software programming language. However, for many high-level programming languages, For bitwise computation-intensive applications, one of such as C/C++, the description of bitwise access and the main difficulties is the lack of bit-accurate computation is not as direct as hardware description descriptions in high-level software programming languages, and hardware synthesis of algorithmic languages. The wide use of bitwise operations in descriptions may generate sub-optimal implement- certain application domains calls for specific bit-level tations for bitwise computation-intensive applications. transformation and optimization to assist hardware In this paper we introduce a bit-level -

Bitwise Operators
Logical operations ANDORNOTXORAND,OR,NOT,XOR •Loggpical operations are the o perations that have its result as a true or false. • The logical operations can be: • Unary operations that has only one operand (NOT) • ex. NOT operand • Binary operations that has two operands (AND,OR,XOR) • ex. operand 1 AND operand 2 operand 1 OR operand 2 operand 1 XOR operand 2 1 Dr.AbuArqoub Logical operations ANDORNOTXORAND,OR,NOT,XOR • Operands of logical operations can be: - operands that have values true or false - operands that have binary digits 0 or 1. (in this case the operations called bitwise operations). • In computer programming ,a bitwise operation operates on one or two bit patterns or binary numerals at the level of their individual bits. 2 Dr.AbuArqoub Truth tables • The following tables (truth tables ) that shows the result of logical operations that operates on values true, false. x y Z=x AND y x y Z=x OR y F F F F F F F T F F T T T F F T F T T T T T T T x y Z=x XOR y x NOT X F F F F T F T T T F T F T T T F 3 Dr.AbuArqoub Bitwise Operations • In computer programming ,a bitwise operation operates on one or two bit patterns or binary numerals at the level of their individual bits. • Bitwise operators • NOT • The bitwise NOT, or complement, is an unary operation that performs logical negation on each bit, forming the ones' complement of the given binary value. -

Parallel Computing
Lecture 1: Single processor performance Why parallel computing • Solving an 푛 × 푛 linear system Ax=b by using Gaussian 1 elimination takes ≈ 푛3 flops. 3 • On Core i7 975 @ 4.0 GHz, which is capable of about 60-70 Gigaflops 푛 flops time 1000 3.3×108 0.006 seconds 1000000 3.3×1017 57.9 days Milestones in Computer Architecture • Analytic engine (mechanical device), 1833 – Forerunner of modern digital computer, Charles Babbage (1792-1871) at University of Cambridge • Electronic Numerical Integrator and Computer (ENIAC), 1946 – Presper Eckert and John Mauchly at the University of Pennsylvania – The first, completely electronic, operational, general-purpose analytical calculator. 30 tons, 72 square meters, 200KW. – Read in 120 cards per minute, Addition took 200µs, Division took 6 ms. • IAS machine, 1952 – John von Neumann at Princeton’s Institute of Advanced Studies (IAS) – Program could be represented in digit form in the computer memory, along with data. Arithmetic could be implemented using binary numbers – Most current machines use this design • Transistors was invented at Bell Labs in 1948 by J. Bardeen, W. Brattain and W. Shockley. • PDP-1, 1960, DEC – First minicomputer (transistorized computer) • PDP-8, 1965, DEC – A single bus (omnibus) connecting CPU, Memory, Terminal, Paper tape I/O and Other I/O. • 7094, 1962, IBM – Scientific computing machine in early 1960s. • 8080, 1974, Intel – First general-purpose 8-bit computer on a chip • IBM PC, 1981 – Started modern personal computer era Remark: see also http://www.computerhistory.org/timeline/?year=1946 -

The P4 Language Specification, V 1.1.0
The P4 Language Specification Version 1.1.0 January 27, 2016 The P4 Language Consortium © 2014-2016, The P4 Language Consortium CONTENTS CONTENTS Contents 1 Introduction5 1.1 The P4 Abstract Model...............................5 1.2 The mTag Example.................................7 1.3 Specification Conventions.............................7 2 Structure of the P4 Language8 2.1 Abstractions.....................................8 2.2 Value Specifications................................9 2.3 Types and declarations............................... 11 2.4 P4 data types..................................... 12 2.4.1 Principles.................................. 12 2.4.2 Base types.................................. 13 2.4.3 Portability.................................. 13 2.4.4 No saturated types............................. 14 2.4.5 Boolean................................... 14 2.4.6 Unsigned integers (bit-strings)...................... 14 2.4.7 Signed Integers............................... 15 2.4.8 Dynamically-sized bit-strings....................... 15 2.4.9 Infinite-precision integers......................... 16 2.4.10 Integer literal types............................. 16 2.5 Base type operations................................ 17 2.5.1 Computations on Boolean values.................... 17 2.5.2 Operations on unsigned fixed-width integers............. 18 2.5.3 Operations on signed fixed-width integers............... 18 2.5.4 A note about shifts............................. 19 2.5.5 varbit operations............................. 21 2.5.6 Operations