A Mathematical Model of the Neuromuscular Junction and Muscle Force Generation in the Pathological Condition Myasthenia Gravis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Neural Control of Movement: Motor Neuron Subtypes, Proprioception and Recurrent Inhibition
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Mem- ic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ, Kullander K (2010) Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as mark- ers for fast motor neurons and partition cells. J Comp Neurol 518:2284-2304. II Wootz H, Enjin A, Wallen-Mackenzie Å, Lindholm D, Kul- lander K (2010) Reduced VGLUT2 expression increases motor neuron viability in Sod1G93A mice. Neurobiol Dis 37:58-66 III Enjin A, Leao KE, Mikulovic S, Le Merre P, Tourtellotte WG, Kullander K. 5-ht1d marks gamma motor neurons and regulates development of sensorimotor connections Manuscript IV Enjin A, Leao KE, Eriksson A, Larhammar M, Gezelius H, Lamotte d’Incamps B, Nagaraja C, Kullander K. Development of spinal motor circuits in the absence of VIAAT-mediated Renshaw cell signaling Manuscript Reprints were made with permission from the respective publishers. Cover illustration Carousel by Sasha Svensson Contents Introduction.....................................................................................................9 Background...................................................................................................11 Neural control of movement.....................................................................11 The motor neuron.....................................................................................12 Organization -

ALS and Other Motor Neuron Diseases Can Represent Diagnostic Challenges
Review Article Address correspondence to Dr Ezgi Tiryaki, Hennepin ALS and Other Motor County Medical Center, Department of Neurology, 701 Park Avenue P5-200, Neuron Diseases Minneapolis, MN 55415, [email protected]. Ezgi Tiryaki, MD; Holli A. Horak, MD, FAAN Relationship Disclosure: Dr Tiryaki’s institution receives support from The ALS Association. Dr Horak’s ABSTRACT institution receives a grant from the Centers for Disease Purpose of Review: This review describes the most common motor neuron disease, Control and Prevention. ALS. It discusses the diagnosis and evaluation of ALS and the current understanding of its Unlabeled Use of pathophysiology, including new genetic underpinnings of the disease. This article also Products/Investigational covers other motor neuron diseases, reviews how to distinguish them from ALS, and Use Disclosure: Drs Tiryaki and Horak discuss discusses their pathophysiology. the unlabeled use of various Recent Findings: In this article, the spectrum of cognitive involvement in ALS, new concepts drugs for the symptomatic about protein synthesis pathology in the etiology of ALS, and new genetic associations will be management of ALS. * 2014, American Academy covered. This concept has changed over the past 3 to 4 years with the discovery of new of Neurology. genes and genetic processes that may trigger the disease. As of 2014, two-thirds of familial ALS and 10% of sporadic ALS can be explained by genetics. TAR DNA binding protein 43 kDa (TDP-43), for instance, has been shown to cause frontotemporal dementia as well as some cases of familial ALS, and is associated with frontotemporal dysfunction in ALS. Summary: The anterior horn cells control all voluntary movement: motor activity, res- piratory, speech, and swallowing functions are dependent upon signals from the anterior horn cells. -

A System for Studying Mechanisms of Neuromuscular Junction Development and Maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2464-2477 doi:10.1242/dev.130278 TECHNIQUES AND RESOURCES RESEARCH ARTICLE A system for studying mechanisms of neuromuscular junction development and maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R. Gomes1,3,*,‡ ABSTRACT different animal models and cell lines (Chen et al., 2014; Corti et al., The neuromuscular junction (NMJ), a cellular synapse between a 2012; Lenzi et al., 2015) with the hope of recapitulating some motor neuron and a skeletal muscle fiber, enables the translation of features of neuromuscular diseases and understanding the triggers chemical cues into physical activity. The development of this special of one of their common hallmarks: the disruption of the structure has been subject to numerous investigations, but its neuromuscular junction (NMJ). The NMJ is one of the most complexity renders in vivo studies particularly difficult to perform. studied synapses. It is formed of three key elements: the lower motor In vitro modeling of the neuromuscular junction represents a powerful neuron (the pre-synaptic compartment), the skeletal muscle (the tool to delineate fully the fine tuning of events that lead to subcellular post-synaptic compartment) and the Schwann cell (Sanes and specialization at the pre-synaptic and post-synaptic sites. Here, we Lichtman, 1999). The NMJ is formed in a step-wise manner describe a novel heterologous co-culture in vitro method using rat following a series of cues involving these three cellular components spinal cord explants with dorsal root ganglia and murine primary and its role is basically to ensure the skeletal muscle functionality. -

The Histology of the Neuromuscular Junction In
75 THE HISTOLOGY OF THE NEUROMUSCULAR JUNCTION Downloaded from https://academic.oup.com/brain/article/84/1/75/372729 by guest on 27 September 2021 IN DYSTROPHIA MYOTONICA BY VIOLET MACDERMOT Department of Neurology, St. Thomas' Hospital, London, S.E.I (1) INTRODUCTION DYSTROPJHC MYOTONICA is a familial disease affecting males and females, usually presenting in adult life, characterized by muscular wasting and weakness together with certain other features. The muscles mainly in- volved are the temporal, masseter, facial, sternomastoid and limb muscles, in the latter those mainly affected being peripheral in distribution. A widespread disorder of muscular contraction, myotonia, is also present but is noticed chiefly in the tongue and in the muscles involved in grasping. The other features of the condition are some degree of mental defect, dysphonia, cataracts, frontal baldness, sparse body hair and testicular atrophy. Any of the manifestations of the disease may be absent and the order of presentation of symptoms is variable. The myotonia may precede muscular wasting by many years or may occur independently. In those muscles which are severely wasted the myotonia tends to disappear. The interest of dystrophia myotonica lies in the peculiar distribution of muscle involvement and in the combination of a disorder of muscle function with endocrine and other dysplasic features. The results of histological examination of biopsy and post-mortem material have been described and reviewed by numerous workers, notably Steinert (1909), Adie and Greenfield (1923), Keschner and Davison (1933), Hassin and Kesert (1948), Wohlfart (1951), Adams, Denny-Brown and Pearson (1953), Greenfield, Shy, Alvord and Berg (1957). -

Lancl1 Promotes Motor Neuron Survival and Extends the Lifespan of Amyotrophic Lateral Sclerosis Mice
Cell Death & Differentiation (2020) 27:1369–1382 https://doi.org/10.1038/s41418-019-0422-6 ARTICLE LanCL1 promotes motor neuron survival and extends the lifespan of amyotrophic lateral sclerosis mice 1 1 1 1 1 1 1 Honglin Tan ● Mina Chen ● Dejiang Pang ● Xiaoqiang Xia ● Chongyangzi Du ● Wanchun Yang ● Yiyuan Cui ● 1,2 1 1,3 4 4 3 1,5 Chao Huang ● Wanxiang Jiang ● Dandan Bi ● Chunyu Li ● Huifang Shang ● Paul F. Worley ● Bo Xiao Received: 19 November 2018 / Revised: 3 September 2019 / Accepted: 6 September 2019 / Published online: 30 September 2019 © The Author(s) 2019. This article is published with open access Abstract Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of motor neurons. Improving neuronal survival in ALS remains a significant challenge. Previously, we identified Lanthionine synthetase C-like protein 1 (LanCL1) as a neuronal antioxidant defense gene, the genetic deletion of which causes apoptotic neurodegeneration in the brain. Here, we report in vivo data using the transgenic SOD1G93A mouse model of ALS indicating that CNS-specific expression of LanCL1 transgene extends lifespan, delays disease onset, decelerates symptomatic progression, and improves motor performance of SOD1G93A mice. Conversely, CNS-specific deletion of fl 1234567890();,: 1234567890();,: LanCL1 leads to neurodegenerative phenotypes, including motor neuron loss, neuroin ammation, and oxidative damage. Analysis reveals that LanCL1 is a positive regulator of AKT activity, and LanCL1 overexpression restores the impaired AKT activity in ALS model mice. These findings indicate that LanCL1 regulates neuronal survival through an alternative mechanism, and suggest a new therapeutic target in ALS. -
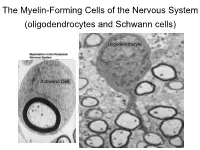
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Cortex Brainstem Spinal Cord Thalamus Cerebellum Basal Ganglia
Harvard-MIT Division of Health Sciences and Technology HST.131: Introduction to Neuroscience Course Director: Dr. David Corey Motor Systems I 1 Emad Eskandar, MD Motor Systems I - Muscles & Spinal Cord Introduction Normal motor function requires the coordination of multiple inter-elated areas of the CNS. Understanding the contributions of these areas to generating movements and the disturbances that arise from their pathology are important challenges for the clinician and the scientist. Despite the importance of diseases that cause disorders of movement, the precise function of many of these areas is not completely clear. The main constituents of the motor system are the cortex, basal ganglia, cerebellum, brainstem, and spinal cord. Cortex Basal Ganglia Cerebellum Thalamus Brainstem Spinal Cord In very broad terms, cortical motor areas initiate voluntary movements. The cortex projects to the spinal cord directly, through the corticospinal tract - also known as the pyramidal tract, or indirectly through relay areas in the brain stem. The cortical output is modified by two parallel but separate re entrant side loops. One loop involves the basal ganglia while the other loop involves the cerebellum. The final outputs for the entire system are the alpha motor neurons of the spinal cord, also called the Lower Motor Neurons. Cortex: Planning and initiation of voluntary movements and integration of inputs from other brain areas. Basal Ganglia: Enforcement of desired movements and suppression of undesired movements. Cerebellum: Timing and precision of fine movements, adjusting ongoing movements, motor learning of skilled tasks Brain Stem: Control of balance and posture, coordination of head, neck and eye movements, motor outflow of cranial nerves Spinal Cord: Spontaneous reflexes, rhythmic movements, motor outflow to body. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Motor Neuron Disease Motor Neuron Disease
Motor Neuron Disease Motor Neuron Disease • Incidence: 2-4 per 100 000 • Onset: usually 50-70 years • Pathology: – Degenerative condition – anterior horn cells and upper motor neurons in spinal cord, resulting in mixed upper and lower motor neuron signs • Cause unknown – 10% familial (SOD-1 mutation) – ? Related to athleticism Presentation • Several variations in onset, but progress to the same endpoint • Motor nerves only affected • May be just UMN or just LMN at onset, but other features will appear over time • Main patterns: – Amyotrophic lateral sclerosis – Bulbar presentaion – Primary lateral sclerosis (UMN onset) – Progressive muscular atrophy (LMN onset) Questions Wasting Classification • Amyotrophic Lateral Sclerosis • Progressive Bulbar Palsy • Progressive Muscular Atrophy • Primary Lateral Sclerosis • Multifocal Motor Neuropathy • Spinal Muscular Atrophy • Kennedy’s Disease • Monomelic Amyotrophy • Brachial Amyotrophic Diplegia El Escorial Criteria for Diagnosis Tongue fasiculations Amyotrophic lateral sclerosis • ‘Typical’ presentation (60%+) • Usually one limb initially – Foot drop – Clumsy weak hand – May complain of cramps • Gradual progression over months • May be some wasting at presentation • Usually fasiculations (often more widespread) • Brisk reflexes, extensor plantars • No sensory signs; MAY occasionally be mild symptoms • Relentless progression, noticable over weeks/ months Bulbar MND • Approximately 30% of cases • Onset with dysarthria, dysphagia • Bulbar and pseudobulbar symptoms • On examination – Dysarthria – -

Olig2 Precursors Produce Abducens Motor Neurons and Oligodendrocytes in the Zebrafish Hindbrain
2322 • The Journal of Neuroscience, February 25, 2009 • 29(8):2322–2333 Cellular/Molecular Olig2ϩ Precursors Produce Abducens Motor Neurons and Oligodendrocytes in the Zebrafish Hindbrain Denise A. Zannino1,2 and Bruce Appel1,3 1Program in Neuroscience and 2Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee 37235, and 3Department of Pediatrics, University of Colorado Denver, Anschutz Medical Campus, Aurora, Colorado 80045 During development, a specific subset of ventral spinal cord precursors called pMN cells produces first motor neurons and then oligo- dendrocyte progenitor cells (OPCs), which migrate, divide and differentiate as myelinating oligodendrocytes. pMN cells express the Olig2 transcription factor and Olig2 function is necessary for formation of spinal motor neurons and OPCs. In the hindbrain and midbrain, distinct classes of visceral, branchiomotor and somatic motor neurons are organized as discrete nuclei, and OPCs are broadly distributed. Mouse embryos deficient for Olig2 function lack somatic motor neurons and OPCs, but it is not clear whether this reflects a common origin for these cells, similar to spinal cord, or independent requirements for Olig2 function in somatic motor neuron and OPC develop- ment. We investigated cranial motor neuron and OPC development in zebrafish and found, using a combination of transgenic reporters and cell type specific antibodies, that somatic abducens motor neurons and a small subset of OPCs arise from common olig2ϩ neuroep- ithelial precursors in rhombomeres r5 and r6, but that all other motor neurons and OPCs do not similarly develop from shared pools of olig2ϩ precursors. In the absence of olig2 function, r5 and r6 precursors remain in the cell cycle and fail to produce abducens motor neurons, and OPCs are entirely lacking in the hindbrain. -

Gamma Motor Neurons Survive and Exacerbate Alpha Motor Neuron Degeneration In
Gamma motor neurons survive and exacerbate alpha PNAS PLUS motor neuron degeneration in ALS Melanie Lalancette-Heberta,b, Aarti Sharmaa,b, Alexander K. Lyashchenkoa,b, and Neil A. Shneidera,b,1 aCenter for Motor Neuron Biology and Disease, Columbia University, New York, NY 10032; and bDepartment of Neurology, Columbia University, New York, NY 10032 Edited by Rob Brownstone, University College London, London, United Kingdom, and accepted by Editorial Board Member Fred H. Gage October 27, 2016 (received for review April 4, 2016) The molecular and cellular basis of selective motor neuron (MN) the muscle spindle and control the sensitivity of spindle afferent vulnerability in amyotrophic lateral sclerosis (ALS) is not known. In discharge (15); beta (β) skeletofusimotor neurons innervate both genetically distinct mouse models of familial ALS expressing intra- and extrafusal muscle (16). In addition to morphological mutant superoxide dismutase-1 (SOD1), TAR DNA-binding protein differences, distinct muscle targets, and the absence of primary 43 (TDP-43), and fused in sarcoma (FUS), we demonstrate selective afferent (IA)inputsonγ-MNs, these functional MN subtypes also degeneration of alpha MNs (α-MNs) and complete sparing of differ in their trophic requirements, and γ-MNs express high levels gamma MNs (γ-MNs), which selectively innervate muscle spindles. of the glial cell line-derived neurotropic factor (GDNF) receptor Resistant γ-MNs are distinct from vulnerable α-MNs in that they Gfrα1 (17). γ-MNs are also molecularly distinguished by the ex- lack synaptic contacts from primary afferent (IA) fibers. Elimination pression of other selective markers including the transcription α of these synapses protects -MNs in the SOD1 mutant, implicating factor Err3 (18), Wnt7A (19), the serotonin receptor 1d (5-ht1d) this excitatory input in MN degeneration. -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function.