The Neuromuscular Junction: Roles in Aging and Neuromuscular Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

This Letter Is for Families with Variant(S) in the Titin Gene, Also
This letter is for families with variant(s) in the Titin gene , also abbreviated as TTN . Changes in the Titin protein may cause muscle weakness as well as heart problems . You will need to discuss with your doctor if and how your Titin variant affects your health. What is Titin? Titin is a very large protein. It’s huge! In fact, Titin is the largest protein in the human body. The Titin protein is located in each of the individual muscle cells in our bodies. It is also found in the heart, which is a very specialized muscle. Muscles need Titin in order to work and move. You can learn more about Titin here: http://titinmyopathy.com . What is a Titin Myopathy? In medical terms, “Myo” refers to muscle and “-opathy” at the end of a word means that the word describes a medical disease or condition. So “myopathy” is a medical illness involving muscles. Myopathies result in muscle weakness and muscle fatigue. “Titin Myopathy” is a specific category of myopathy where the muscle problem is caused by a change in the Titin gene and subsequently the protein. What is a Titin-related Dystrophy? A Titin dystrophy is a muscle disorder where muscle cells break down. Dystrophies generally result in weakness that gets worse over time. A common heart problem caused by variants in the Titin gene is known as dilated cardiomyopathy. Sometimes other heart issues are also present in people with changes in their Titin gene. It is a good idea to have a checkup from a heart doctor if you have even a single variant in the Titin gene. -

A System for Studying Mechanisms of Neuromuscular Junction Development and Maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2464-2477 doi:10.1242/dev.130278 TECHNIQUES AND RESOURCES RESEARCH ARTICLE A system for studying mechanisms of neuromuscular junction development and maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R. Gomes1,3,*,‡ ABSTRACT different animal models and cell lines (Chen et al., 2014; Corti et al., The neuromuscular junction (NMJ), a cellular synapse between a 2012; Lenzi et al., 2015) with the hope of recapitulating some motor neuron and a skeletal muscle fiber, enables the translation of features of neuromuscular diseases and understanding the triggers chemical cues into physical activity. The development of this special of one of their common hallmarks: the disruption of the structure has been subject to numerous investigations, but its neuromuscular junction (NMJ). The NMJ is one of the most complexity renders in vivo studies particularly difficult to perform. studied synapses. It is formed of three key elements: the lower motor In vitro modeling of the neuromuscular junction represents a powerful neuron (the pre-synaptic compartment), the skeletal muscle (the tool to delineate fully the fine tuning of events that lead to subcellular post-synaptic compartment) and the Schwann cell (Sanes and specialization at the pre-synaptic and post-synaptic sites. Here, we Lichtman, 1999). The NMJ is formed in a step-wise manner describe a novel heterologous co-culture in vitro method using rat following a series of cues involving these three cellular components spinal cord explants with dorsal root ganglia and murine primary and its role is basically to ensure the skeletal muscle functionality. -

The Histology of the Neuromuscular Junction In
75 THE HISTOLOGY OF THE NEUROMUSCULAR JUNCTION Downloaded from https://academic.oup.com/brain/article/84/1/75/372729 by guest on 27 September 2021 IN DYSTROPHIA MYOTONICA BY VIOLET MACDERMOT Department of Neurology, St. Thomas' Hospital, London, S.E.I (1) INTRODUCTION DYSTROPJHC MYOTONICA is a familial disease affecting males and females, usually presenting in adult life, characterized by muscular wasting and weakness together with certain other features. The muscles mainly in- volved are the temporal, masseter, facial, sternomastoid and limb muscles, in the latter those mainly affected being peripheral in distribution. A widespread disorder of muscular contraction, myotonia, is also present but is noticed chiefly in the tongue and in the muscles involved in grasping. The other features of the condition are some degree of mental defect, dysphonia, cataracts, frontal baldness, sparse body hair and testicular atrophy. Any of the manifestations of the disease may be absent and the order of presentation of symptoms is variable. The myotonia may precede muscular wasting by many years or may occur independently. In those muscles which are severely wasted the myotonia tends to disappear. The interest of dystrophia myotonica lies in the peculiar distribution of muscle involvement and in the combination of a disorder of muscle function with endocrine and other dysplasic features. The results of histological examination of biopsy and post-mortem material have been described and reviewed by numerous workers, notably Steinert (1909), Adie and Greenfield (1923), Keschner and Davison (1933), Hassin and Kesert (1948), Wohlfart (1951), Adams, Denny-Brown and Pearson (1953), Greenfield, Shy, Alvord and Berg (1957). -

The Myotonic Dystrophies: Diagnosis and Management Chris Turner,1 David Hilton-Jones2
Review J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.2008.158261 on 22 February 2010. Downloaded from The myotonic dystrophies: diagnosis and management Chris Turner,1 David Hilton-Jones2 1Department of Neurology, ABSTRACT asymptomatic relatives as well as prenatal and National Hospital for Neurology There are currently two clinically and molecularly defined preimplantation diagnosis can also be performed.7 and Neurosurgery, London, UK 2Department of Clinical forms of myotonic dystrophy: (1) myotonic dystrophy Neurology, The Radcliffe type 1 (DM1), also known as ‘Steinert’s disease’; and Anticipation Infirmary, Oxford, UK (2) myotonic dystrophy type 2 (DM2), also known as DMPK alleles greater than 37 CTG repeats in length proximal myotonic myopathy. DM1 and DM2 are are unstable and may expand in length during meiosis Correspondence to progressive multisystem genetic disorders with several and mitosis. Children of a parent with DM1 may Dr C Turner, Department of Neurology, National Hospital for clinical and genetic features in common. DM1 is the most inherit repeat lengths considerably longer than those Neurology and Neurosurgery, common form of adult onset muscular dystrophy whereas present in the transmitting parent. This phenomenon Queen Square, London WC1N DM2 tends to have a milder phenotype with later onset of causes ‘anticipation’, which is the occurrence of 3BG, UK; symptoms and is rarer than DM1. This review will focus increasing disease severity and decreasing age of onset [email protected] on the clinical features, diagnosis and management of in successive generations. The presence of a larger Received 1 December 2008 DM1 and DM2 and will briefly discuss the recent repeat leads to earlier onset and more severe disease Accepted 18 December 2008 advances in the understanding of the molecular and causes the more severe phenotype of ‘congenital’ pathogenesis of these diseases with particular reference DM1 (figure 2).8 9 A child with congenital DM 1 to new treatments using gene therapy. -
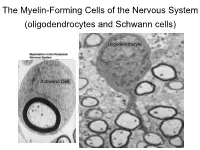
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Skeletal Muscle Channelopathies: a Guide to Diagnosis and Management
Review Pract Neurol: first published as 10.1136/practneurol-2020-002576 on 9 February 2021. Downloaded from Skeletal muscle channelopathies: a guide to diagnosis and management Emma Matthews ,1,2 Sarah Holmes,3 Doreen Fialho2,3,4 1Atkinson- Morley ABSTRACT in the case of myotonia may be precipi- Neuromuscular Centre, St Skeletal muscle channelopathies are a group tated by sudden or initial movement, George's University Hospitals NHS Foundation Trust, London, of rare episodic genetic disorders comprising leading to falls and injury. Symptoms are UK the periodic paralyses and the non- dystrophic also exacerbated by prolonged rest, espe- 2 Department of Neuromuscular myotonias. They may cause significant morbidity, cially after preceding physical activity, and Diseases, UCL, Institute of limit vocational opportunities, be socially changes in environmental temperature.4 Neurology, London, UK 3Queen Square Centre for embarrassing, and sometimes are associated Leg muscle myotonia can cause particular Neuromuscular Diseases, with sudden cardiac death. The diagnosis is problems on public transport, with falls National Hospital for Neurology often hampered by symptoms that patients may caused by the vehicle stopping abruptly and Neurosurgery, London, UK 4Department of Clinical find difficult to describe, a normal examination or missing a destination through being Neurophysiology, King's College in the absence of symptoms, and the need unable to rise and exit quickly enough. Hospital NHS Foundation Trust, to interpret numerous tests that may be These difficulties can limit independence, London, UK normal or abnormal. However, the symptoms social activity, choice of employment Correspondence to respond very well to holistic management and (based on ability both to travel to the Dr Emma Matthews, Atkinson- pharmacological treatment, with great benefit to location and to perform certain tasks) and Morley Neuromuscular Centre, quality of life. -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function. -

Evidence-Based Guideline: Evaluation, Diagnosis, and Management Of
Evidence-based Guideline: Evaluation, Diagnosis, and Management of Facioscapulohumeral Muscular Dystrophy Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine Rabi Tawil, MD, FAAN1; John T. Kissel, MD, FAAN2; Chad Heatwole, MD, MS-CI3; Shree Pandya, PT, DPT, MS4; Gary Gronseth, MD, FAAN5; Michael Benatar, MBChB, DPhil, FAAN6 (1) MDA Neuromuscular Disease Clinic, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY (2) Department of Neurology, Wexner Medical Center, Ohio State University, Columbus, OH (3) Department of Neurology, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY (4) Department of Neurology, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY (5) Department of Neurology, University of Kansas School of Medicine, Kansas City, KS (6) Department of Neurology, Miller School of Medicine, University of Miami, Miami, FL Correspondence to: American Academy of Neurology [email protected] 1 Approved by the Guideline Development, Dissemination, and Implementation Subcommittee on July 23, 2014; by the AAN Practice Committee on October 20, 2014; by the AANEM Board of Directors on [date]; and by the AANI Board of Directors on [date]. This guideline was endorsed by the FSH Society on December 18, 2014. 2 AUTHOR CONTRIBUTIONS Rabi Tawil: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision. John Kissel: acquisition of data, analysis or interpretation of data, critical revision of the manuscript for important intellectual content. -

Myopathies Infosheet
The University of Chicago Genetic Services Laboratories 5841 S. Maryland Ave., Rm. G701, MC 0077, Chicago, Illinois 60637 Toll Free: (888) UC GENES (888) 824 3637 Local: (773) 834 0555 FAX: (773) 702 9130 [email protected] dnatesting.uchicago.edu CLIA #: 14D0917593 CAP #: 18827-49 Gene tic Testing for Congenital Myopathies/Muscular Dystrophies Congenital Myopathies Congenital myopathies are typically characterized by the presence of specific structural and histochemical features on muscle biopsy and clinical presentation can include congenital hypotonia, muscle weakness, delayed motor milestones, feeding difficulties, and facial muscle involvement (1). Serum creatine kinase may be normal or elevated. Heterogeneity in presenting symptoms can occur even amongst affected members of the same family. Congenital myopathies can be divided into three main clinicopathological defined categories: nemaline myopathy, core myopathy and centronuclear myopathy (2). Nemaline Myopathy Nemaline Myopathy is characterized by weakness, hypotonia and depressed or absent deep tendon reflexes. Weakness is typically proximal, diffuse or selective, with or without facial weakness and the diagnostic hallmark is the presence of distinct rod-like inclusions in the sarcoplasm of skeletal muscle fibers (3). Core Myopathy Core Myopathy is characterized by areas lacking histochemical oxidative and glycolytic enzymatic activity on histopathological exam (2). Symptoms include proximal muscle weakness with onset either congenitally or in early childhood. Bulbar and facial weakness may also be present. Patients with core myopathy are typically subclassified as either having central core disease or multiminicore disease. Centronuclear Myopathy Centronuclear Myopathy (CNM) is a rare muscle disease associated with non-progressive or slowly progressive muscle weakness that can develop from infancy to adulthood (4, 5). -

Clinical Approach to the Floppy Child
THE FLOPPY CHILD CLINICAL APPROACH TO THE FLOPPY CHILD The floppy infant syndrome is a well-recognised entity for paediatricians and neonatologists and refers to an infant with generalised hypotonia presenting at birth or in early life. An organised approach is essential when evaluating a floppy infant, as the causes are numerous. A detailed history combined with a full systemic and neurological examination are critical to allow for accurate and precise diagnosis. Diagnosis at an early stage is without a doubt in the child’s best interest. HISTORY The pre-, peri- and postnatal history is important. Enquire about the quality and quantity of fetal movements, breech presentation and the presence of either poly- or oligohydramnios. The incidence of breech presentation is higher in fetuses with neuromuscular disorders as turning requires adequate fetal mobility. Documentation of birth trauma, birth anoxia, delivery complications, low cord R van Toorn pH and Apgar scores are crucial as hypoxic-ischaemic encephalopathy remains MB ChB, (Stell) MRCP (Lond), FCP (SA) an important cause of neonatal hypotonia. Neonatal seizures and an encephalo- Specialist pathic state offer further proof that the hypotonia is of central origin. The onset of the hypotonia is also important as it may distinguish between congenital and Department of Paediatrics and Child Health aquired aetiologies. Enquire about consanguinity and identify other affected fam- Faculty of Health Sciences ily members in order to reach a definitive diagnosis, using a detailed family Stellenbosch University and pedigree to assist future genetic counselling. Tygerberg Children’s Hospital CLINICAL CLUES ON NEUROLOGICAL EXAMINATION Ronald van Toorn obtained his medical degree from the University of Stellenbosch, There are two approaches to the diagnostic problem. -

Neuromuscular Junctions LEARNING OBJECTIVES: ➢ Components of the Neuromuscular Junction (NMJ) ➢ Physiological Anatomy of NMJ
NEUROMUSCULAR JUNCTION By:Dr.Fareeda banu A.B. Associate professor Dept of Physiology, USM-KLE IMP Neuromuscular Junctions LEARNING OBJECTIVES: ➢ Components of the neuromuscular junction (NMJ) ➢ Physiological anatomy of NMJ. ➢ Synthesis, storage and release of Ach at NMJ ➢ Events occurring during neuromuscular transmission with reference to end-plate potentials. ➢ Clinical importance of NMJ ➢ Drugs affecting NMJ transmission. ➢ Applied aspects. INTRODUCTION Movement of the body requires the interaction of a complex series of afferent and efferent neuronal signals, which result in a skeletal muscle contraction The neuromuscular junction is a specialized form of a chemical synapse comprised of an alpha motor neuron and the muscle fiber it innervates. INTRODUCTION Junctions Vrs. Synapses - NMJ is a junction between a motor neuron and a skeletal muscle. Synapse is a connection between two neurons A junction will always respond to an action potential in the presynaptic nerve. Synapses may or may not. A junction will have a safety factor, sufficient release of neurotransmitter to insure action potential generation in the effector organ, usually a thousand or so times over. MOTOR UNIT: THE NERVE-MUSCLE FUNCTIONAL UNIT ❖ Motor Unit: The Nerve-Muscle Functional Unit. A motor unit is a motor neuron and all the muscle fibers it innervate ❖ The nerve fiber forms a complex of branching nerve terminals that invaginate into the surface of the muscle fiber but lie outside the muscle fiber plasma membrane. The entire structure is called the motor end plate. NEUROMUSCULAR JUNCTION (NMJ) Defn: NMJ is a junction between the motor nerve ending and skeletal muscle fiber. Components of the NMJ: It is comprised of; Unmyelinated terminal boutons of axon supplying a skeletal muscle fiber.