Measuring and Modeling of Axon Membrane Properties in Motor Neuron Disorders and Normal Subjects
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1960 Surname
Surname Given Age Date Page Maiden Note Abbett Marda R. 25-Jan A-11 Abel Maude 53 4-Apr B-3 Abercrombie Julia 63 8-Nov A-11 Acheson Robert Worth 63 23-Aug B-3 Acker Ella 88 28-Mar B-3 Adamchuk Steve 65 30-Aug A-11 Adamek Anna 86 4-Sep B-3 Adams Helen B. 49 15-Jul B-3 Adams Homer Taylor 75 21-Mar B-3 Addlesberger Frank H. 62 14-Jun B-3 Adelsperger Carolina C. (Carrie)_ 69 18-Nov B-3 Adlers Nellie C.. 43 14-Feb B-3 Aguilar Juan O., Jr. 19 24-Feb 1 Ahedo Lupe 62 17-Aug B-3 Ahlendorf Alvina L. 74 4-Aug B-3 Ahrendt Martha 70 28-Dec B-3 Ainsworth Alta Belle 76 28-Jul A-11 Albertsen Rosella 61 22-Feb A-11 Alexander Ernest R. 83 14-Nov B-3 Alexander Joseph H. 82 15-Jul B-3 Allande Emil 54 17-Jun B-3 Allen William 14-Jun B-3 Alley Margaret B. 53 18-Jan A-11 Almanzia Maria 72 3-Oct B-3 Alvarado Ruby 49 11-Jan A-11 Alvey Wylie G. 80 19-Sep A-11 Ambre Henry L. 85 7-Nov B-5 Ambrose Paul R. 2 26-May 1 Andel Michael, Sr. 69 14-Sep B-3 Andersen Neils P. 74 13-Jun B-3 Anderson Daniel 2 19-Jan A-9 Anderson Donald R. 47 3-Jul B-3 Anderson Irene 59 4-Dec B-3 Anderson Jessie (Rohde) 66 18-Jan A-11 Anderson John B. -

IN TAX LEADERS WOMEN in TAX LEADERS | 4 AMERICAS Latin America
WOMEN IN TAX LEADERS THECOMPREHENSIVEGUIDE TO THE WORLD’S LEADING FEMALE TAX ADVISERS SIXTH EDITION IN ASSOCIATION WITH PUBLISHED BY WWW.INTERNATIONALTAXREVIEW.COM Contents 2 Introduction and methodology 8 Bouverie Street, London EC4Y 8AX, UK AMERICAS Tel: +44 20 7779 8308 4 Latin America: 30 Costa Rica Fax: +44 20 7779 8500 regional interview 30 Curaçao 8 United States: 30 Guatemala Editor, World Tax and World TP regional interview 30 Honduras Jonathan Moore 19 Argentina 31 Mexico Researchers 20 Brazil 31 Panama Lovy Mazodila 24 Canada 31 Peru Annabelle Thorpe 29 Chile 32 United States Jason Howard 30 Colombia 41 Venezuela Production editor ASIA-PACIFIC João Fernandes 43 Asia-Pacific: regional 58 Malaysia interview 59 New Zealand Business development team 52 Australia 60 Philippines Margaret Varela-Christie 53 Cambodia 61 Singapore Raquel Ipo 54 China 61 South Korea Managing director, LMG Research 55 Hong Kong SAR 62 Taiwan Tom St. Denis 56 India 62 Thailand 58 Indonesia 62 Vietnam © Euromoney Trading Limited, 2020. The copyright of all 58 Japan editorial matter appearing in this Review is reserved by the publisher. EUROPE, MIDDLE EAST & AFRICA 64 Africa: regional 101 Lithuania No matter contained herein may be reproduced, duplicated interview 101 Luxembourg or copied by any means without the prior consent of the 68 Central Europe: 102 Malta: Q&A holder of the copyright, requests for which should be regional interview 105 Malta addressed to the publisher. Although Euromoney Trading 72 Northern & 107 Netherlands Limited has made every effort to ensure the accuracy of this Southern Europe: 110 Norway publication, neither it nor any contributor can accept any regional interview 111 Poland legal responsibility whatsoever for consequences that may 86 Austria 112 Portugal arise from errors or omissions, or any opinions or advice 87 Belgium 115 Qatar given. -

Šiauliai University Faculty of Humanities Department of English Philology
ŠIAULIAI UNIVERSITY FACULTY OF HUMANITIES DEPARTMENT OF ENGLISH PHILOLOGY RENDERING OF GERMANIC PROPER NAMES IN THE LITHUANIAN PRESS BACHELOR THESIS Research Adviser: Assist. L.Petrulion ė Student: Aist ė Andži ūtė Šiauliai, 2010 CONTENTS INTRODUCTION......................................................................................................................3 1. THE CONCEPTION OF PROPER NAMES.........................................................................5 1.2. The development of surnames.............................................................................................6 1.3. Proper names in Germanic languages .................................................................................8 1.3.1. Danish, Norwegian, Swedish and Icelandic surnames.................................................9 1.3.2. Dutch surnames ..........................................................................................................12 1.3.3. English surnames........................................................................................................13 1.3.4. German surnames .......................................................................................................14 2. NON-LITHUANIAN SURNAMES ORTHOGRAPHY .....................................................16 2.1. The historical development of the problem.......................................................................16 2.2. The rules of transcriptions of non-Lithuanian proper names ............................................22 3. THE USAGE -
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -

Characterization of Visceral and Subcutaneous Adipose Tissue
J. Perinat. Med. 2016; 44(7): 813–835 Shali Mazaki-Tovi*, Adi L. Tarca, Edi Vaisbuch, Juan Pedro Kusanovic, Nandor Gabor Than, Tinnakorn Chaiworapongsa, Zhong Dong, Sonia S. Hassan and Roberto Romero* Characterization of visceral and subcutaneous adipose tissue transcriptome in pregnant women with and without spontaneous labor at term: implication of alternative splicing in the metabolic adaptations of adipose tissue to parturition DOI 10.1515/jpm-2015-0259 groups (unpaired analyses) and adipose tissue regions Received July 27, 2015. Accepted October 26, 2015. Previously (paired analyses). Selected genes were tested by quantita- published online March 19, 2016. tive reverse transcription-polymerase chain reaction. Abstract Results: Four hundred and eighty-two genes were differ- entially expressed between visceral and subcutaneous Objective: The aim of this study was to determine gene fat of pregnant women with spontaneous labor at term expression and splicing changes associated with par- (q-value < 0.1; fold change > 1.5). Biological processes turition and regions (visceral vs. subcutaneous) of the enriched in this comparison included tissue and vascu- adipose tissue of pregnant women. lature development as well as inflammatory and meta- Study design: The transcriptome of visceral and abdomi- bolic pathways. Differential splicing was found for 42 nal subcutaneous adipose tissue from pregnant women at genes [q-value < 0.1; differences in Finding Isoforms using term with (n = 15) and without (n = 25) spontaneous labor Robust Multichip Analysis scores > 2] between adipose was profiled with the Affymetrix GeneChip Human Exon tissue regions of women not in labor. Differential exon 1.0 ST array. Overall gene expression changes and the dif- usage associated with parturition was found for three ferential exon usage rate were compared between patient genes (LIMS1, HSPA5, and GSTK1) in subcutaneous tissues. -
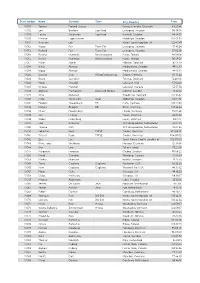
Start Number Name Surname Team City, Country Time 11001 Tommy
Start number Name Surname Team City, Country Time 11001 Tommy Frølund Jensen Kongens lyngby, Denmark 89:20:46 11002 Emil Boström Lag Nord Linköping, Sweden 98:19:04 11003 Lovisa Johansson Lag Nord Burträsk, Sweden 98:19:02 11005 Maurice Lagershausen Huddinge, Sweden 102:01:54 11011 Hyun jun Lee Korea, (south) republic of 122:40:49 11012 Viggo Fält Team Fält Linköping, Sweden 57:45:24 11013 Richard Fält Team Fält Linköping, Sweden 57:45:28 11014 Rasmus Nummela Rackmannarna Pjelax, Finland 98:59:06 11015 Rafael Nummela Rackmannarna Pjelax, Finland 98:59:06 11017 Marie Zølner Hillerød, Denmark 82:11:49 11018 Niklas Åkesson Mellbystrand, Sweden 99:45:22 11019 Viggo Åkesson Mellbystrand, Sweden 99:45:22 11020 Damian Sulik #StopComplaining Siegen, Germany 101:13:24 11023 Bruno Svendsen Tylstrup, Denmark 74:48:08 11024 Philip Oswald Lakeland, USA 55:52:02 11025 Virginia Marshall Lakefield, Canada 55:57:54 11026 Andreas Mathiasson Backyard Heroes Ödsmål, Sweden 75:12:02 11028 Stine Andersen Brædstrup, Denmark 104:35:37 11030 Patte Johansson Tiger Södertälje, Sweden 80:24:51 11031 Frederic Wiesenbach BB Dahn, Germany 101:21:50 11032 Romea Brugger BB Berlin, Germany 101:22:44 11034 Oliver Freudenberg Heyda, Germany 75:07:48 11035 Jan Hennig Farum, Denmark 82:11:50 11036 Robert Falkenberg Farum, Denmark 82:11:51 11037 Edo Boorsma Sint annaparochie, Netherlands 123:17:34 11038 Trijneke Stuit Sint annaparochie, Netherlands 123:17:35 11040 Sebastian Keck TATSE Gieäen, Germany 104:08:01 11041 Tatjana Kage TATSE Gieäen, Germany 104:08:02 11042 Eun Lee -

BELGIUM NAME № YEAR Abbeloos, Antoine & Flore (Devos) 944 1975 Abras, Joseph & Jeanne (Delage) 12468 2012 Aelbers, Mathieu H
Righteous Among the Nations Honored by Yad Vashem by 1 January 2020 BELGIUM NAME № YEAR Abbeloos, Antoine & Flore (Devos) 944 1975 Abras, Joseph & Jeanne (Delage) 12468 2012 Aelbers, Mathieu H. & Maria (Alberts) 2794 1984 Aendekerk, Petrus Johannes H. 3109 1985 Aerts Jean 10604 2005 Alardo, Victor & Ida; ch: Marcelle, Marie-Louise, 5483 1992 Joseph Alleman, Jean-Baptiste & Augusta 6978 1996 Amelot, Adele 7357 1996 Anciaux, Emile & Josephine 6466 1994 Andre Emile & daughter Madeleine 8864 2000 Andre Emilie & sister Juliette 8864.1 2000 Andre, Joseph 486 1968 Andre, Paul & Jeanne 2407 1983 Andries, Joseph Jean & Marie (Vanden Bergh) 13158 2015 Andriesse-Renard, Paule 7065 1996 Aneuse, Nelly (Mother Julienne Aneuse) 13034.3 2015 Annaert, Petrus & Isabella, her mother Catherine 11130.1 2007 Steenput-Jacobs Ansiaux, Nicolas J. & Marie Celine 4031.1 1988 Antonis-Maluin, Rene & Leontine (Depraetere) 4435 1989 Arcq, Georgees & Lydie 8432 1999 Arekens, Marie 7465.1 1997 Arnauts, Marie T.E. (Mere Ursule) 11793 2010 Arnoldy, Marcel & Celestine (Schyns) 11350 2008 Arnould, Fernand & Nelly (Bayard) 756 1972 Artus, Mathilde 3127 1985 Audenardt, Pieter & Victorine & daughter Germaine 5986 1994 (Van 't Hoff) Avaux, Evence & Irene (Grimee) 8306 1998 Baeck, Bernard & Arlette (Peeters) 11697 2009 Baeten, Antoon & Maria (Jaspers) 1748 1980 Baggen, Helene 9938 2003 Baichez, Fernand & Juliette-Marie 1638.4 2002 Bal, Auguste & Odile 8998 2000 Bamps, Louis & Elisa 9981 2003 Bams, Joannes & Elvira (Borgugnons) 13402 2017 Bartholomeus, Urbain & Marie-Louise 8910 -

Human Genetics and Clinical Aspects of Neurodevelopmental Disorders
bioRxiv preprint doi: https://doi.org/10.1101/000687; this version posted October 6, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Human genetics and clinical aspects of neurodevelopmental disorders Gholson J. Lyon1,2,3,*, Jason O’Rawe1,3 1Stanley Institute for Cognitive Genomics, One Bungtown Road, Cold Spring Harbor Laboratory, NY, USA, 11724 2Institute for Genomic Medicine, Utah Foundation for Biomedical Research, E 3300 S, Salt Lake City, Salt Lake City, UT, USA, 84106 3Stony Brook University, 100 Nicolls Rd, Stony Brook, NY, USA, 11794 * Corresponding author: Gholson J. Lyon Email: [email protected] Other author emails: Jason O'Rawe: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/000687; this version posted October 6, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Introduction “our incomplete studies do not permit actual classification; but it is better to leave things by themselves rather than to force them into classes which have their foundation only on paper” – Edouard Seguin (Seguin, 1866) “The fundamental mistake which vitiates all work based upon Mendel’s method is the neglect of ancestry, and the attempt to regard the whole effect upon off- spring, produced by a particular parent, as due to the existence in the parent of particular structural characters; while the contradictory results obtained by those who have observed the offspring of parents apparently identical in cer- tain characters show clearly enough that not only the parents themselves, but their race, that is their ancestry, must be taken into account before the result of pairing them can be predicted” – Walter Frank Raphael Weldon (Weldon, 1902). -

Immunogenicity 28 and 35 Has Multiple Effects on T Cell Type I IFN
Type I IFN Induced by Adenovirus Serotypes 28 and 35 Has Multiple Effects on T Cell Immunogenicity This information is current as Matthew J. Johnson, Constantinos Petrovas, Takuya of September 24, 2021. Yamamoto, Ross W. B. Lindsay, Karin Loré, Jason G. D. Gall, Emma Gostick, François Lefebvre, Mark J. Cameron, David A. Price, Elias Haddad, Rafick-Pierre Sekaly, Robert A. Seder and Richard A. Koup J Immunol 2012; 188:6109-6118; Prepublished online 14 Downloaded from May 2012; doi: 10.4049/jimmunol.1103717 http://www.jimmunol.org/content/188/12/6109 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2012/05/14/jimmunol.110371 Material 7.DC1 References This article cites 66 articles, 37 of which you can access for free at: http://www.jimmunol.org/content/188/12/6109.full#ref-list-1 Why The JI? Submit online. by guest on September 24, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Type I IFN Induced by Adenovirus Serotypes 28 and 35 Has Multiple Effects on T Cell Immunogenicity Matthew J. -

Exploration, Disruption, Diaspora: Movement of Nuevomexicanos to Utah, 1776-1850 Linda C
University of New Mexico UNM Digital Repository American Studies ETDs Electronic Theses and Dissertations Spring 5-11-2019 Exploration, Disruption, Diaspora: Movement of Nuevomexicanos to Utah, 1776-1850 Linda C. Eleshuk Roybal Follow this and additional works at: https://digitalrepository.unm.edu/amst_etds Part of the American Studies Commons Recommended Citation Eleshuk Roybal, Linda C.. "Exploration, Disruption, Diaspora: Movement of Nuevomexicanos to Utah, 1776-1850." (2019). https://digitalrepository.unm.edu/amst_etds/80 This Dissertation is brought to you for free and open access by the Electronic Theses and Dissertations at UNM Digital Repository. It has been accepted for inclusion in American Studies ETDs by an authorized administrator of UNM Digital Repository. For more information, please contact [email protected]. Linda Catherine Eleshuk Roybal Candidate American Studies Department This dissertation is approved and it is acceptable in quality and form for publication: Approved by the Dissertation Committee: A.Gabriel Meléndez, Chair Kathleen Holscher Michelle Hall Kells Enrique Lamadrid i Exploration, Disruption, Diaspora: Movement Of Nuevomexicanos to Utah, 1776 – 1950 By Linda Catherine Eleshuk Roybal B.S. Psychology, Weber State College, 1973 B.S. Communications, Weber State University, 1982 M.S. English, Utah State University, 1997 DISSERTATION Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy In American Studies The University of New Mexico Albuquerque, New Mexico May 2019 ii Dedication This dissertation is dedicated to my children and grandchildren, born and yet to be — Joys of my life and ambassadors to a future I will not see, Con mucho cariño. iii Acknowledgements I am grateful to so many people who have shared their expertise and resources to bring this project to its completion. -

GOO-80-02119 392P
DOCUMENT RESUME ED 228 863 FL 013 634 AUTHOR Hatfield, Deborah H.; And Others TITLE A Survey of Materials for the Study of theUncommonly Taught Languages: Supplement, 1976-1981. INSTITUTION Center for Applied Linguistics, Washington, D.C. SPONS AGENCY Department of Education, Washington, D.C.Div. of International Education. PUB DATE Jul 82 CONTRACT GOO-79-03415; GOO-80-02119 NOTE 392p.; For related documents, see ED 130 537-538, ED 132 833-835, ED 132 860, and ED 166 949-950. PUB TYPE Reference Materials Bibliographies (131) EDRS PRICE MF01/PC16 Plus Postage. DESCRIPTORS Annotated Bibliographies; Dictionaries; *InStructional Materials; Postsecondary Edtmation; *Second Language Instruction; Textbooks; *Uncommonly Taught Languages ABSTRACT This annotated bibliography is a supplement tothe previous survey published in 1976. It coverslanguages and language groups in the following divisions:(1) Western Europe/Pidgins and Creoles (European-based); (2) Eastern Europeand the Soviet Union; (3) the Middle East and North Africa; (4) SouthAsia;(5) Eastern Asia; (6) Sub-Saharan Africa; (7) SoutheastAsia and the Pacific; and (8) North, Central, and South Anerica. The primaryemphasis of the bibliography is on materials for the use of theadult learner whose native language is English. Under each languageheading, the items are arranged as follows:teaching materials, readers, grammars, and dictionaries. The annotations are descriptive.Whenever possible, each entry contains standardbibliographical information, including notations about reprints and accompanyingtapes/records -

Growth of Malignant Extracranial Tumors Alters Micrornaome in the Prefrontal Cortex of Tumorgraft Mice
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No.51), pp: 88276-88293 Research Paper: Gerotarget (Focus on Aging) Growth of malignant extracranial tumors alters microRNAome in the prefrontal cortex of TumorGraft mice Anna Kovalchuk1,4, Yaroslav Ilnytskyy2, Rocio Rodriguez-Juarez2, Amanda Katz3, David Sidransky3, Bryan Kolb1 and Olga Kovalchuk2 1 Department of Neuroscience, University of Lethbridge, Lethbridge, AB, Canada 2 Department of Biological Sciences, University of Lethbridge, Lethbridge, AB, Canada 3 Department of Oncology, Champions Oncology, Baltimore, MD, USA 4 Leaders in Medicine Program, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada Correspondence to: Bryan Kolb, email: [email protected] Correspondence to:Olga Kovalchuk, email: [email protected] Keywords: chemo brain, tumor brain, epigenetics, microRNA, Gerotarget Received: April 04, 2017 Accepted: April 27, 2017 Published: August 03, 2017 Copyright: Kovalchuk et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License 3.0 (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT A wide array of central nervous system complications, neurological deficits, and cognitive impairments occur and persist as a result of systemic cancer and cancer treatments. This condition is known as chemo brain and it affects over half of cancer survivors. Recent studies reported that cognitive impairments manifest before chemotherapy and are much broader than chemo brain alone, thereby adding in tumor brain as a component. The molecular mechanisms of chemo brain are under- investigated, and the mechanisms of tumor brain have not been analyzed at all.