Omics, Epigenetics, and Genome Editing Techniques for Food and Nutritional Security
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
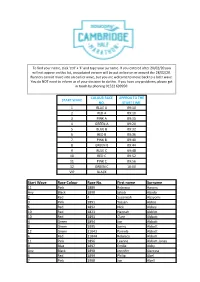
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

Listado De Personas Admitidas
PRUEBA PARA LA ELABORACIÓN DE LISTAS DE CONTRATACIÓN TEMPORAL DE ESPECIALISTA DE APOYO EDUCATIVO A través de una prueba selectiva, se constituirán dos relaciones de aspirantes, una en castellano y otra en euskera, para optar a puestos de Especialista de Apoyo Educativo en centros educativos, dependientes del Departamento de Educación. ASISTENCIA A LA PRUEBA. ELECCIÓN DE ZONAS Y JORNADA Las personas que aparecen en el listado facilitado por el SNE, deberán cumplimentar un formulario disponible, a través del botón “Tramitar” en el siguiente enlace para indicar tres aspectos obligatorios para la gestión de las personas aspirantes: 1. Confirmar su asistencia el día de la prueba. 2. Seleccionar las zonas en la que se quiere figurar para las ofertas de contratación. 3. Indicar el tipo de jornada por la que se quiere optar para las ofertas de contratación. El plazo para enviar el formulario cumplimentado, es de diez días hábiles, del 23 de febrero al 8 de marzo de 2021, ambos incluidos. FECHA DE REALIZACIÓN DE LA PRUEBA La prueba se celebrará en Pamplona el sábado 27 de marzo de 2021, por la mañana. Por la situación sanitaria actual debido a la COVID-19, una vez se conozca el número de personas que confirmen su asistencia el día de la prueba, se publicará el lugar de realización de la misma y el protocolo a seguir para cumplir con todas las normas sanitarias establecidas. HEZKUNTZA-LAGUNTZAKO ESPEZIALISTAK ALDI BATERAKO KONTRATATZEKO ZERRENDAK EGITEKO PROBA Hautaproba baten bidez, izangaien bi zerrenda eratuko dira, bata gaztelaniaz eta bestea euskaraz, Hezkuntza Sailaren mendeko ikastetxeetan Hezkuntza Laguntzako Espezialista lanpostuak lortzeko. -

IN TAX LEADERS WOMEN in TAX LEADERS | 4 AMERICAS Latin America
WOMEN IN TAX LEADERS THECOMPREHENSIVEGUIDE TO THE WORLD’S LEADING FEMALE TAX ADVISERS SIXTH EDITION IN ASSOCIATION WITH PUBLISHED BY WWW.INTERNATIONALTAXREVIEW.COM Contents 2 Introduction and methodology 8 Bouverie Street, London EC4Y 8AX, UK AMERICAS Tel: +44 20 7779 8308 4 Latin America: 30 Costa Rica Fax: +44 20 7779 8500 regional interview 30 Curaçao 8 United States: 30 Guatemala Editor, World Tax and World TP regional interview 30 Honduras Jonathan Moore 19 Argentina 31 Mexico Researchers 20 Brazil 31 Panama Lovy Mazodila 24 Canada 31 Peru Annabelle Thorpe 29 Chile 32 United States Jason Howard 30 Colombia 41 Venezuela Production editor ASIA-PACIFIC João Fernandes 43 Asia-Pacific: regional 58 Malaysia interview 59 New Zealand Business development team 52 Australia 60 Philippines Margaret Varela-Christie 53 Cambodia 61 Singapore Raquel Ipo 54 China 61 South Korea Managing director, LMG Research 55 Hong Kong SAR 62 Taiwan Tom St. Denis 56 India 62 Thailand 58 Indonesia 62 Vietnam © Euromoney Trading Limited, 2020. The copyright of all 58 Japan editorial matter appearing in this Review is reserved by the publisher. EUROPE, MIDDLE EAST & AFRICA 64 Africa: regional 101 Lithuania No matter contained herein may be reproduced, duplicated interview 101 Luxembourg or copied by any means without the prior consent of the 68 Central Europe: 102 Malta: Q&A holder of the copyright, requests for which should be regional interview 105 Malta addressed to the publisher. Although Euromoney Trading 72 Northern & 107 Netherlands Limited has made every effort to ensure the accuracy of this Southern Europe: 110 Norway publication, neither it nor any contributor can accept any regional interview 111 Poland legal responsibility whatsoever for consequences that may 86 Austria 112 Portugal arise from errors or omissions, or any opinions or advice 87 Belgium 115 Qatar given. -

Integrative Analysis for Elucidating Transcriptomics Landscapes of Glucocorticoid-Induced Osteoporosis
fcell-08-00252 April 13, 2020 Time: 17:59 # 1 ORIGINAL RESEARCH published: 16 April 2020 doi: 10.3389/fcell.2020.00252 Integrative Analysis for Elucidating Transcriptomics Landscapes of Glucocorticoid-Induced Osteoporosis Xiaoxia Ying1†, Xiyun Jin2†, Pingping Wang2†, Yuzhu He1, Haomiao Zhang1, Xiang Ren1, Songling Chai1, Wenqi Fu1, Pengcheng Zhao1, Chen Chen1, Guowu Ma1* and Huiying Liu1* 1 2 Edited by: School of Stomatology, Dalian Medical University, Dalian, China, School of Life Sciences and Technology, Harbin Institute Yongchun Zuo, of Technology, Harbin, China Inner Mongolia University, China Reviewed by: Osteoporosis is the most common bone metabolic disease, characterized by bone Liang Yu, mass loss and bone microstructure changes due to unbalanced bone conversion, Xidian University, China Yanshuo Chu, which increases bone fragility and fracture risk. Glucocorticoids are clinically used to The University of Texas MD Anderson treat a variety of diseases, including inflammation, cancer and autoimmune diseases. Cancer Center, United States However, excess glucocorticoids can cause osteoporosis. Herein we performed an *Correspondence: Guowu Ma integrated analysis of two glucocorticoid-related microarray datasets. The WGCNA [email protected] analysis identified 3 and 4 glucocorticoid-related gene modules, respectively. Differential Huiying Liu expression analysis revealed 1047 and 844 differentially expressed genes in the two [email protected] datasets. After integrating differentially expressed glucocorticoid-related genes, we †These authors have contributed equally to this work found that most of the robust differentially expressed genes were up-regulated. Through protein-protein interaction analysis, we obtained 158 glucocorticoid-related candidate Specialty section: This article was submitted to genes. Enrichment analysis showed that these genes are significantly enriched in the Epigenomics and Epigenetics, osteoporosis related pathways. -

The ELIXIR Core Data Resources: Fundamental Infrastructure for The
Supplementary Data: The ELIXIR Core Data Resources: fundamental infrastructure for the life sciences The “Supporting Material” referred to within this Supplementary Data can be found in the Supporting.Material.CDR.infrastructure file, DOI: 10.5281/zenodo.2625247 (https://zenodo.org/record/2625247). Figure 1. Scale of the Core Data Resources Table S1. Data from which Figure 1 is derived: Year 2013 2014 2015 2016 2017 Data entries 765881651 997794559 1726529931 1853429002 2715599247 Monthly user/IP addresses 1700660 2109586 2413724 2502617 2867265 FTEs 270 292.65 295.65 289.7 311.2 Figure 1 includes data from the following Core Data Resources: ArrayExpress, BRENDA, CATH, ChEBI, ChEMBL, EGA, ENA, Ensembl, Ensembl Genomes, EuropePMC, HPA, IntAct /MINT , InterPro, PDBe, PRIDE, SILVA, STRING, UniProt ● Note that Ensembl’s compute infrastructure physically relocated in 2016, so “Users/IP address” data are not available for that year. In this case, the 2015 numbers were rolled forward to 2016. ● Note that STRING makes only minor releases in 2014 and 2016, in that the interactions are re-computed, but the number of “Data entries” remains unchanged. The major releases that change the number of “Data entries” happened in 2013 and 2015. So, for “Data entries” , the number for 2013 was rolled forward to 2014, and the number for 2015 was rolled forward to 2016. The ELIXIR Core Data Resources: fundamental infrastructure for the life sciences 1 Figure 2: Usage of Core Data Resources in research The following steps were taken: 1. API calls were run on open access full text articles in Europe PMC to identify articles that mention Core Data Resource by name or include specific data record accession numbers. -

Ecological Developmental Biology and Disease States CHAPTER 5 Teratogenesis: Environmental Assaults on Development 167
Integrating Epigenetics, Medicine, and Evolution Scott F. Gilbert David Epel Swarthmore College Hopkins Marine Station, Stanford University Sinauer Associates, Inc. • Publishers Sunderland, Massachusetts U.S.A. © Sinauer Associates, Inc. This material cannot be copied, reproduced, manufactured or disseminated in any form without express written permission from the publisher. Brief Contents PART 1 Environmental Signals and Normal Development CHAPTER 1 The Environment as a Normal Agent in Producing Phenotypes 3 CHAPTER 2 How Agents in the Environment Effect Molecular Changes in Development 37 CHAPTER 3 Developmental Symbiosis: Co-Development as a Strategy for Life 79 CHAPTER 4 Embryonic Defenses: Survival in a Hostile World 119 PART 2 Ecological Developmental Biology and Disease States CHAPTER 5 Teratogenesis: Environmental Assaults on Development 167 CHAPTER 6 Endocrine Disruptors 197 CHAPTER 7 The Epigenetic Origin of Adult Diseases 245 PART 3 Toward a Developmental Evolutionary Synthesis CHAPTER 8 The Modern Synthesis: Natural Selection of Allelic Variation 289 CHAPTER 9 Evolution through Developmental Regulatory Genes 323 CHAPTER 10 Environment, Development, and Evolution: Toward a New Synthesis 369 CODA Philosophical Concerns Raised by Ecological Developmental Biology 403 APPENDIX A Lysenko, Kammerer, and the Truncated Tradition of Ecological Developmental Biology 421 APPENDIX B The Molecular Mechanisms of Epigenetic Change 433 APPENDIX C Writing Development Out of the Modern Synthesis 441 APPENDIX D Epigenetic Inheritance Systems: -
![[3 TD$DIFF]Interdisciplinary Team Science in Cell Biology](https://docslib.b-cdn.net/cover/7372/3-td-diff-interdisciplinary-team-science-in-cell-biology-237372.webp)
[3 TD$DIFF]Interdisciplinary Team Science in Cell Biology
TICB 1268 No. of Pages 3 Scientific Life Cell biology, beginning largely as micro- detailed physical–chemical mechanisms Interdisciplinary[3_TD$IF] scopic observations, followed[1_TD$IF]the molec- [7]. The data required for these models ular biology revolution, which viewed are now in sight. New gene editing meth- Team Science in genes, cells, and the machinery that ods are providing endogenous expression underlies their activities as molecular sys- of tagged and mutant cells [8], and new Cell Biology tems that could be fully characterized and live-cell imaging methods are promising Rick Horwitz1,* understood using methods of genetics biochemistry in living cells, measuring con- and biochemistry. Viewing the cell as a centrations, dynamics, equilibria, and complex, dynamic molecular composite organization [9]. Similarly, super-resolution The cell is complex. With its multi- brought insights from chemistry and phys- microscopy and cryoEM tomography, tude of components, spatial– ics to bear on biological problems. Just as which allow structure determination and [6_TD$IF] temporal character, and gene the molecular genetic era was codified by organization in situ [3,4], imaging mass expression diversity, it is challeng- the publication of Watson's book, Molec- spectrometry [10], and single-cell and ing to comprehend the cell as an ular Biology of the Gene [1], two decades spatially-resolved genomic approaches integrated system and to develop later[8_TD$IF]the Molecular Biology of the Cell by [11–13], among other image-based tech- models that predict its behaviors. I Alberts, et al. [2] served a similar purpose nologies, all point to a new golden era of suggest an approach to address for cell biology. -

DNA Sequencing and Sorting: Identifying Genetic Variations
BioMath DNA Sequencing and Sorting: Identifying Genetic Variations Student Edition Funded by the National Science Foundation, Proposal No. ESI-06-28091 This material was prepared with the support of the National Science Foundation. However, any opinions, findings, conclusions, and/or recommendations herein are those of the authors and do not necessarily reflect the views of the NSF. At the time of publishing, all included URLs were checked and active. We make every effort to make sure all links stay active, but we cannot make any guaranties that they will remain so. If you find a URL that is inactive, please inform us at [email protected]. DIMACS Published by COMAP, Inc. in conjunction with DIMACS, Rutgers University. ©2015 COMAP, Inc. Printed in the U.S.A. COMAP, Inc. 175 Middlesex Turnpike, Suite 3B Bedford, MA 01730 www.comap.com ISBN: 1 933223 71 5 Front Cover Photograph: EPA GULF BREEZE LABORATORY, PATHO-BIOLOGY LAB. LINDA SHARP ASSISTANT This work is in the public domain in the United States because it is a work prepared by an officer or employee of the United States Government as part of that person’s official duties. DNA Sequencing and Sorting: Identifying Genetic Variations Overview Each of the cells in your body contains a copy of your genetic inheritance, your DNA which has been passed down to you, one half from your biological mother and one half from your biological father. This DNA determines physical features, like eye color and hair color, and can determine susceptibility to medical conditions like hypertension, heart disease, diabetes, and cancer. -

Early-Life Experience, Epigenetics, and the Developing Brain
Neuropsychopharmacology REVIEWS (2015) 40, 141–153 & 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 REVIEW ............................................................................................................................................................... www.neuropsychopharmacologyreviews.org 141 Early-Life Experience, Epigenetics, and the Developing Brain 1 ,1 Marija Kundakovic and Frances A Champagne* 1Department of Psychology, Columbia University, New York, NY, USA Development is a dynamic process that involves interplay between genes and the environment. In mammals, the quality of the postnatal environment is shaped by parent–offspring interactions that promote growth and survival and can lead to divergent developmental trajectories with implications for later-life neurobiological and behavioral characteristics. Emerging evidence suggests that epigenetic factors (ie, DNA methylation, posttranslational histone modifications, and small non- coding RNAs) may have a critical role in these parental care effects. Although this evidence is drawn primarily from rodent studies, there is increasing support for these effects in humans. Through these molecular mechanisms, variation in risk of psychopathology may emerge, particularly as a consequence of early-life neglect and abuse. Here we will highlight evidence of dynamic epigenetic changes in the developing brain in response to variation in the quality of postnatal parent–offspring interactions. The recruitment of epigenetic pathways for the -

Men Final Entries
Final Entries - Athletes List by event European Athletics Indoor Championships 2021, Torun/POL Tot. Number of countries Tot. Number of athletes Tot. Number of Men Tot. Number of Women 47 733 405 328 FINAL ENTRIES - Men 60m Senior Men Num. of countries: 33 Num. of athletes: 71 Member Federation Surname First Name DoB PB SB ARM Donigian Alexander 20/10/1993 6.64i 6.79i ART Keletela Dorian Celeste 06/02/1999 6.79i 6.85i AUT Fuchs Markus 14/11/1995 6.62i 6.69i BEL Kuba Di-Vita Gaylord 17/11/1995 6.73i 6.75i BEL Vleminckx Kobe 31/05/1998 6.65i 6.65i BLR Bliznets Dzianis 12/03/1995 6.75i 6.75i BLR Bohdan Maksim 19/03/1997 6.77i 6.77i BLR Zabalotny Yury 24/02/1997 6.72i 6.72i BUL Dimitrov Denis 10/02/1994 6.65i 6.73i BUL Jivkov Vesselin 26/01/2001 6.76i 6.80i CZE Hampl Štěpán 10/11/1999 6.70i 6.70i CZE Stromšík Zdeněk 25/11/1994 6.60i 6.68i CZE Veleba Jan 06/12/1986 6.65i 6.65i DEN Hansen Simon 30/06/1998 6.75i 6.75i DEN Kjær Emil Mader 20/12/1999 6.77i 6.77i DEN Musah Kojo 15/04/1996 6.61i 6.61i ESP López Sergio 05/07/1999 6.67i 6.74i ESP Rodríguez Daniel 26/01/1995 6.67i 6.67i ESP Sanchez Ricardo 10/08/1999 6.75i 6.75i EST Nazarov Karl Erik 17/03/1999 6.63i 6.63i FIN Illukka Riku 21/09/1999 6.73i 6.73i FIN Purola Samuel 19/05/2000 6.67i 6.67i FIN Samuelsson Samuli 23/06/1995 6.66i 6.66i FRA Fall Mouhamadou 25/02/1992 6.62i 6.62i FRA Golitin Amaury 28/01/1997 6.62i 6.62i GBR Aikines-Aryeetey Harry 29/08/1988 6.55i 6.67i GBR Bromby Oliver 30/03/1998 6.63i 6.65i GBR Robertson Andrew 17/12/1990 6.54i 6.61i GER Corucle Philipp 18/07/1997 6.62i -

EPIGENOMICS: BEYOND Cpg ISLANDS
REVIEWS EPIGENOMICS: BEYOND CpG ISLANDS Melissa J. Fazzari* and John M. Greally† Epigenomic studies aim to define the location and nature of the genomic sequences that are epigenetically modified. Much progress has been made towards whole-genome epigenetic profiling using molecular techniques, but the analysis of such large and complex data sets is far from trivial given the correlated nature of sequence and functional characteristics within the genome. We describe the statistical solutions that help to overcome the problems with data-set complexity, in anticipation of the imminent wealth of data that will be generated by new genome- wide epigenetic profiling and DNA sequence analysis techniques. So far, epigenomic studies have succeeded in identifying CpG islands, but recent evidence points towards a role for transposable elements in epigenetic regulation, causing the fields of study of epigenetics and transposable element biology to converge. Epigenetic inheritance involves the transmission of issues of correlation and causality — for example, the information not encoded in DNA sequences from cell DNA sequence feature might be the effect of the epige- to daughter cell or from generation to generation. netic process rather than mechanistically involved in Covalent modifications of the DNA or its packaging directing it. As new techniques to characterize epigenetic histones are responsible for transmitting epigenetic processes throughout the genome are being applied, we information. Epigenomics can be defined as a genome- have the potential to generate large amounts of data to wide approach to studying epigenetics. This term facilitate epigenomic studies. It is a good time now to encompasses whole-genome studies of epigenetic consider these issues so that we can design our analytical processes and the identification of the DNA sequences approaches appropriately. -

Genomic Divergence and Brain Evolution: How Regulatory DNA Influences Development of the Cerebral Cortex
Prospects & Overviews Review essays Genomic divergence and brain evolution: How regulatory DNA influences development of the cerebral cortex Debra L. Silver1)2)3)4) The cerebral cortex controls our most distinguishing higher Introduction cognitive functions. Human-specific gene expression dif- ferences are abundant in the cerebral cortex, yet we have A large six-layered neocortex is a unique feature of only begun to understand how these variations impact brain mammalian brains. This specialized outer covering of the brain controls our higher cognitive functions including function. This review discusses the current evidence linking abstract thought and language, which together help uniquely non-coding regulatory DNA changes, including enhancers, define us as humans. Our distinguishing cognitive capacities with neocortical evolution. Functional interrogation using are specified within discrete cortical areas and are driven by animal models reveals converging roles for our genome in dynamic communication between neurons of the neocortex key aspects of cortical development including progenitor and other brain regions, as well as glial cell populations (including oligodendrocytes, microglia, and astrocytes). cell cycle and neuronal signaling. New technologies, Neurons are initially generated during human embryonic includingiPS cells and organoids, offerpotential alternatives and early fetal development, where they migrate to appropri- to modeling evolutionary modifications in a relevant species ate regions and begin establishing functional connections context. Several diseases rooted in the cerebral cortex during fetal and postnatal stages (Fig. 1). Disruptions to uniquely manifest in humans compared to other primates, cerebral cortex function arising during either development or thus highlighting the importance of understanding human adulthood, can result in neurodevelopmental and neurode- generative disorders.