Neurology, Neuromuscular, and Cardiology Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Program Nr: 1 from the 2004 ASHG Annual Meeting Mutations in A
Program Nr: 1 from the 2004 ASHG Annual Meeting Mutations in a novel member of the chromodomain gene family cause CHARGE syndrome. L.E.L.M. Vissers1, C.M.A. van Ravenswaaij1, R. Admiraal2, J.A. Hurst3, B.B.A. de Vries1, I.M. Janssen1, W.A. van der Vliet1, E.H.L.P.G. Huys1, P.J. de Jong4, B.C.J. Hamel1, E.F.P.M. Schoenmakers1, H.G. Brunner1, A. Geurts van Kessel1, J.A. Veltman1. 1) Dept Human Genetics, UMC Nijmegen, Nijmegen, Netherlands; 2) Dept Otorhinolaryngology, UMC Nijmegen, Nijmegen, Netherlands; 3) Dept Clinical Genetics, The Churchill Hospital, Oxford, United Kingdom; 4) Children's Hospital Oakland Research Institute, BACPAC Resources, Oakland, CA. CHARGE association denotes the non-random occurrence of ocular coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies and deafness (OMIM #214800). Almost all patients with CHARGE association are sporadic and its cause was unknown. We and others hypothesized that CHARGE association is due to a genomic microdeletion or to a mutation in a gene affecting early embryonic development. In this study array- based comparative genomic hybridization (array CGH) was used to screen patients with CHARGE association for submicroscopic DNA copy number alterations. De novo overlapping microdeletions in 8q12 were identified in two patients on a genome-wide 1 Mb resolution BAC array. A 2.3 Mb region of deletion overlap was defined using a tiling resolution chromosome 8 microarray. Sequence analysis of genes residing within this critical region revealed mutations in the CHD7 gene in 10 of the 17 CHARGE patients without microdeletions, including 7 heterozygous stop-codon mutations. -

Kabuki Syndrome
Kabuki syndrome Description Kabuki syndrome is a disorder that affects many parts of the body. It is characterized by distinctive facial features including arched eyebrows; long eyelashes; long openings of the eyelids (long palpebral fissures) with the lower lids turned out (everted) at the outside edges; a flat, broadened tip of the nose; and large protruding earlobes. The name of this disorder comes from the resemblance of its characteristic facial appearance to stage makeup used in traditional Japanese Kabuki theater. People with Kabuki syndrome have mild to severe developmental delay and intellectual disability. Affected individuals may also have seizures, an unusually small head size ( microcephaly), or weak muscle tone (hypotonia). Some have eye problems such as rapid, involuntary eye movements (nystagmus) or eyes that do not look in the same direction (strabismus). Other characteristic features of Kabuki syndrome include short stature and skeletal abnormalities such as abnormal side-to-side curvature of the spine (scoliosis), short fifth (pinky) fingers, or problems with the hip and knee joints. The roof of the mouth may have an abnormal opening (cleft palate) or be high and arched, and dental problems are common in affected individuals. People with Kabuki syndrome may also have fingerprints with unusual features and fleshy pads at the tips of the fingers. These prominent finger pads are called fetal finger pads because they normally occur in human fetuses; in most people they disappear before birth. A wide variety of other health problems occur in some people with Kabuki syndrome. Among the most commonly reported are heart abnormalities, frequent ear infections ( otitis media), hearing loss, and early puberty. -

General Contribution
24 Abstracts of 37th Annual Meeting A1 A SCREENING METHOD FOR FRAGILE X MUTATION: DETECTION OF THE CGG REPEAT IN FMR-1 GENE BY PCR WITH BIOTIN-LABELED PRIMER. ..Eiji NANBA, Kousaku OHNO and Kenzo TAKESHITA Division of Child Neurology, Institute of Neurological Sciences, Tot- tori University School of Medicine. Yonago We have developed a new polymerase chain reaction(PCR)-based method for detection of the CGG repeat in FMR-1 gene. No specific product from PCR was detected on the gel with ethidium bromide staining, because 7-deaza-2'-dGTP is necessary for amplification of this repeat. Biotin-labeled primer was used for PCR and the product was transferred to a nylon membrane followed the detection of biotin by Smilight kit. The size of PCR product from normal control were slightly various and around 300bp. No PCR product was detected from 3 fragile X male patients in 2 families diagnosed by cytogenetic examination. This method is useful for genetic screen- ing of male mental retardation patients to exclude the fragile X mutation. A2 DNA ANALYSISFOR FRAGILE X SYNDROME Osamu KOSUDA,Utak00GASA, ~.ideynki INH, a~ji K/NAGIJCltI, and Kazumasa ]tIKIJI (SILL Inc., Tokyo) Fragile X syndrome is X-linked disease having the amplification of (CG6)n repeat sequence in the chromsomeXq27.3. We performed Southern blot analysis using three probes recognized repetitive sequence resion. Normal controle showed 5.2Kb with Eco RI digest and 2.7Kb with Eco RI/Bss ttII digest as the germ tines by the Southern blot analysis. However, three cell lines established fro~ unrelated the patients with fragile X showed the abnormal bands between 5.2 and 7.7Kb with Eco RI digest, and between 2.7 and 7.7Kb with Eco aI/Bss HII digest. -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Hereditary Interstitial Lung Diseases Manifesting in Early Childhood in Japan
nature publishing group Clinical Investigation Articles Hereditary interstitial lung diseases manifesting in early childhood in Japan Takuma Akimoto1, Kazutoshi Cho1, Itaru Hayasaka1, Keita Morioka1, Yosuke Kaneshi1, Itsuko Furuta2, Masafumi Yamada3, Tadashi Ariga3 and Hisanori Minakami2 BACKGROUND: Genetic variations associated with intersti- (SP)-B deficiency (1), SP-C abnormality (2), and ATP-binding tial lung diseases (ILD) have not been extensively studied in cassette A3 (ABCA3) deficiency (3). Disorder of alveolar mac- Japanese infants. rophages includes abnormality in granulocyte macrophage METHODS: Forty-three infants with unexplained lung dys- colony–stimulating factor (GM-CSF) receptor (4,5) and dys- function were studied. All 43, 22, and 17 infants underwent function of macrophages associated with hypogammaglobu- analyses of surfactant protein (SP)-C gene (SFTPC) and ATP- linemia (6). Although considerable overlapping exists, genetic binding cassette A3 gene (ABCA3), SP-B gene (SFTPB), and disorders of SP-B and alveolar macrophages are likely to mani- SP-B western blotting, respectively. Two and four underwent fest hereditary pulmonary alveolar proteinosis (hPAP), while assessment of granulocyte macrophage colony-stimulating those of SP-C and ABCA3 are likely to manifest hPAP and/or factor–stimulating phosphorylation of signal transducer and interstitial pneumonitis (7). In addition, genetic disorders of activator of transcription-5 (pSTAT-5) and analyses of FOXF1 thyroid transcription factor-1–associated thyroid dysfunction gene (FOXF1), respectively. (8) and alveolar capillary dysplasia with misalignment of pul- RESULTS: ILD were diagnosed clinically in nine infants: four, monary veins (ACD/MPV) (9) often manifest interstitial lung three, and two had interstitial pneumonitis, hereditary pul- disease (ILD) in early childhood. -

X-Linked Diseases: Susceptible Females
REVIEW ARTICLE X-linked diseases: susceptible females Barbara R. Migeon, MD 1 The role of X-inactivation is often ignored as a prime cause of sex data include reasons why women are often protected from the differences in disease. Yet, the way males and females express their deleterious variants carried on their X chromosome, and the factors X-linked genes has a major role in the dissimilar phenotypes that that render women susceptible in some instances. underlie many rare and common disorders, such as intellectual deficiency, epilepsy, congenital abnormalities, and diseases of the Genetics in Medicine (2020) 22:1156–1174; https://doi.org/10.1038/s41436- heart, blood, skin, muscle, and bones. Summarized here are many 020-0779-4 examples of the different presentations in males and females. Other INTRODUCTION SEX DIFFERENCES ARE DUE TO X-INACTIVATION Sex differences in human disease are usually attributed to The sex differences in the effect of X-linked pathologic variants sex specific life experiences, and sex hormones that is due to our method of X chromosome dosage compensation, influence the function of susceptible genes throughout the called X-inactivation;9 humans and most placental mammals – genome.1 5 Such factors do account for some dissimilarities. compensate for the sex difference in number of X chromosomes However, a major cause of sex-determined expression of (that is, XX females versus XY males) by transcribing only one disease has to do with differences in how males and females of the two female X chromosomes. X-inactivation silences all X transcribe their gene-rich human X chromosomes, which is chromosomes but one; therefore, both males and females have a often underappreciated as a cause of sex differences in single active X.10,11 disease.6 Males are the usual ones affected by X-linked For 46 XY males, that X is the only one they have; it always pathogenic variants.6 Females are biologically superior; a comes from their mother, as fathers contribute their Y female usually has no disease, or much less severe disease chromosome. -

SUPPLEMENTARY MATERIAL Effect of Next
SUPPLEMENTARY MATERIAL Effect of Next-Generation Exome Sequencing Depth for Discovery of Diagnostic Variants KKyung Kim1,2,3†, Moon-Woo Seong4†, Won-Hyong Chung3, Sung Sup Park4, Sangseob Leem1, Won Park5,6, Jihyun Kim1,2, KiYoung Lee1,2*‡, Rae Woong Park1,2* and Namshin Kim5,6** 1Department of Biomedical Informatics, Ajou University School of Medicine, Suwon 443-749, Korea 2Department of Biomedical Science, Graduate School, Ajou University, Suwon 443-749, Korea, 3Korean Bioinformation Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea, 4Department of Laboratory Medicine, Seoul National University Hospital College of Medicine, Seoul 110-799, Korea, 5Department of Functional Genomics, Korea University of Science and Technology, Daejeon 305-806, Korea, 6Epigenomics Research Center, Genome Institute, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea http//www. genominfo.org/src/sm/gni-13-31-s001.pdf Supplementary Table 1. List of diagnostic genes Gene Symbol Description Associated diseases ABCB11 ATP-binding cassette, sub-family B (MDR/TAP), member 11 Intrahepatic cholestasis ABCD1 ATP-binding cassette, sub-family D (ALD), member 1 Adrenoleukodystrophy ACVR1 Activin A receptor, type I Fibrodysplasia ossificans progressiva AGL Amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase Glycogen storage disease ALB Albumin Analbuminaemia APC Adenomatous polyposis coli Adenomatous polyposis coli APOE Apolipoprotein E Apolipoprotein E deficiency AR Androgen receptor Androgen insensitivity -

Opthalmic Genetics
Inherited Genetics Opthalmic Genetics What is Opthalmic Genetics? Developmental • Anterior segment dysgenesis Of the approximately 5000 genetic diseases and syndromes known to affect humans, at • Aniridia least one-third involve the eye. Due to advances in molecular genetics and sequencing • Anophthlamos/ microphthalmos/ nanophthalmos methods, there has been an exponential increase in the knowledge of genetic eye diseases and syndromes. • Coloboma Complex Prevalence • Cataract • Keratoconus • More than 60% of cases of blindness among infants are caused by inherited eye • Fuchs endothelial corneal dystrophy diseases such as congenital cataracts, congenital glaucoma, retinal degeneration, • Age-related macular degeneration optic atrophy, eye malformations and corneal dystrophies. • Glaucoma • Pseudoexfoliation syndrome What are the common Genetic Opthalmic • Diabetic retinopathy Disorders ? Why do you need to test for Genetic Ophthalmic Disorders can be classified according to the type of genetic abnormality Opthalmic Disorders? Monogenic • There is also evidence now that the most common vision problems among children and adults are genetically determined (Eg: strabismus, amblyopia, refractive errors • Corneal dystrophies such as myopia, hyperopia and astigmatism) • Oculocutaneous Albinism • Genetic ophthalmic disorders include a large number of ocular pathologies which • Norrie disease have autosomal dominant, autosomal recessive or X-linked inheritance patterns, or • Retinoschisis are complex traits with polygenic and environmental components -

Blueprint Genetics Congenital Structural Heart Disease Panel
Congenital Structural Heart Disease Panel Test code: CA1501 Is a 114 gene panel that includes assessment of non-coding variants. Is ideal for patients with congenital heart disease, particularly those with features of hereditary disorders. Is not ideal for patients suspected to have a ciliopathy or a rasopathy. For those patients, please consider our Primary Ciliary Dyskinesia Panel and our Noonan Syndrome Panel, respectively. About Congenital Structural Heart Disease There are many types of congenital heart disease (CHD) ranging from simple asymptomatic defects to complex defects with severe, life-threatening symptoms. CHDs are the most common type of birth defect and affect at least 8 out of every 1,000 newborns. Annually, more than 35,000 babies in the United States are born with CHDs. Many of these CHDs are simple conditions and need no treatment or are easily repaired. Some babies are born with complex CHD requiring special medical care. The diagnosis and treatment of complex CHDs has greatly improved over the past few decades. As a result, almost all children who have complex heart defects survive to adulthood and can live active, productive lives. However, many patients who have complex CHDs continue to need special heart care throughout their lives. In the United States, more than 1 million adults are living with congenital heart disease. Availability 4 weeks Gene Set Description Genes in the Congenital Structural Heart Disease Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ABL1 Congenital -

Genetic Investigation of 211 Chinese Families Expands the Mutational and Phenotypical Spectrum in Hereditary Retinopathy Genes Through Targeted Sequencing Technology
Genetic investigation of 211 Chinese families expands the mutational and phenotypical spectrum in hereditary retinopathy genes through targeted sequencing technology Zhouxian Bai The First Aliated Hospital of Zhengzhou University https://orcid.org/0000-0001-7071-666X Yanchuan Xie First Aliated Hospital of Henan University of Science and technology Lina Liu First Aliated Hospital of Zhengzhou University Jingzhi Shao Southern Medical University Nanfang Hospital Yuying Liu First Aliated Hospital of Zhengzhou University Xiangdong Kong ( [email protected] ) https://orcid.org/0000-0003-0030-7638 Research article Keywords: hereditary retinopathy, novel mutations, targeted sequencing, genetic testing Posted Date: December 4th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-20958/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at BMC Medical Genomics on March 29th, 2021. See the published version at https://doi.org/10.1186/s12920-021-00935-w. Page 1/22 Abstract Background: Hereditary retinopathy is a signicant cause of blindness worldwide. Despite the discovery of many mutations in various retinopathies, a large part of patients remain undiagnosed genetically. Targeted next generation sequencing of the human genome is a suitable approach for retinopathy molecular diagnosis. Methods: We described a cohort of 211 families from central China with various forms of retinopathy, 95 families of which were investigated using NGS multi-gene panel sequencing as well as the other 116 patients were LHON suspected tested by Sanger sequencing. We validated the candidate variants by PCR- based Sanger sequencing. We have made comprehensive analysis of the cases through sequencing data and ophthalmologic examination information. -
ISCA Disease List
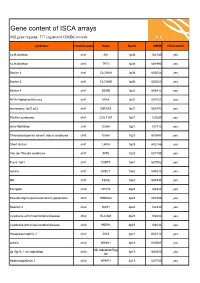
Gene content of ISCA arrays 402 gene regions, 377 registered OMIM records syndrome chromosome Gene Band OMIM ISCA 8x60k 1p36 deletion chr1 SKI 1p36 164780 yes 1p36 deletion chr1 TP73 1p36 601990 yes Bartter 4 chr1 CLCNKA 1p36 602024 yes Bartter 3 chr1 CLCNKB 1p36 602023 yes Bartter 4 chr1 BSND 1p32 606412 yes NFIA Haploinsufficiency chr1 NFIA 1p31 600727 yes monosomy 1p31 p22 chr1 DIRAS3 1p31 605193 yes Stickler syndrome chr1 COL11A1 1p21 120280 yes atrial fibrillation chr1 GJA5 1q21 121013 yes Thrombocytopenia absent radius syndrome chr1 GJA8 1q21 600897 yes Short stature chr1 LHX4 1q25 602146 yes Van der Woude syndrome chr1 IRF6 1q32 607199 yes Fryns 1q41 chr1 DISP1 1q41 607502 yes autism chr1 DISC1 1q42 605210 yes MR chr1 TBCE 1q42 604934 yes Feingold chr2 MYCN 2q24 164840 yes Pseudovaginal perineoscrotal hypospadias chr2 SRD5A2 2p23 607306 yes Noonan 4 chr2 SOS1 2p22 182530 yes Cystinuria with mitochondrial disease chr2 SLC3A1 2p21 104614 yes Cystinuria with mitochondrial disease chr2 PREPL 2p21 104614 yes Holopresencaphly 2 chr2 SIX3 2p21 603714 yes autism chr2 NRXN1 2p16 600565 yes MicrodeletionReg 2p15p16.1 microdeletion chr2 2p15 602559 yes ion Nephronophthsis 1 chr2 NPHP1 2q13 607100 yes Holoprosencephaly 9 chr2 GLI2 2q14 165230 yes visceral heterotaxy chr2 CFC1 2q21 605194 yes Mowat-Wilson syndrome chr2 ZEB2 2q22 605802 yes autism chr2 SLC4A10 2q24 605556 yes SCN1A-related seizures chr2 SCN1A 2q24 182389 yes HYPOMYELINATION, GLOBAL CEREBRAL chr2 SLC25A12 2q31 603667 yes Split/hand foot malformation -5 chr2 DLX1 2q31 600029 yes -
ORD Resources Report
Resources and their URL's 12/1/2013 Resource Name: Resource URL: 1 in 9: The Long Island Breast Cancer Action Coalition http://www.1in9.org 11q Research and Resource Group http://www.11qusa.org 1p36 Deletion Support & Awareness http://www.1p36dsa.org 22q11 Ireland http://www.22q11ireland.org 22qcentral.org http://22qcentral.org 2q23.org http://2q23.org/ 4p- Support Group http://www.4p-supportgroup.org/ 4th Angel Mentoring Program http://www.4thangel.org 5p- Society http://www.fivepminus.org A Foundation Building Strength http://www.buildingstrength.org A National Support group for Arthrogryposis Multiplex http://www.avenuesforamc.com Congenita (AVENUES) A Place to Remember http://www.aplacetoremember.com/ Aarons Ohtahara http://www.ohtahara.org/ About Special Kids http://www.aboutspecialkids.org/ AboutFace International http://aboutface.ca/ AboutFace USA http://www.aboutfaceusa.org Accelerate Brain Cancer Cure http://www.abc2.org Accelerated Cure Project for Multiple Sclerosis http://www.acceleratedcure.org Accord Alliance http://www.accordalliance.org/ Achalasia 101 http://achalasia.us/ Acid Maltase Deficiency Association (AMDA) http://www.amda-pompe.org Acoustic Neuroma Association http://anausa.org/ Addison's Disease Self Help Group http://www.addisons.org.uk/ Adenoid Cystic Carcinoma Organization International http://www.accoi.org/ Adenoid Cystic Carcinoma Research Foundation http://www.accrf.org/ Advocacy for Neuroacanthocytosis Patients http://www.naadvocacy.org Advocacy for Patients with Chronic Illness, Inc. http://www.advocacyforpatients.org