Opthalmic Genetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Defects of CHM and Visual Acuity Outcome in 24 Choroideremia
www.nature.com/scientificreports OPEN Genetic defects of CHM and visual acuity outcome in 24 choroideremia patients from 16 Japanese families Takaaki Hayashi1,2*, Shuhei Kameya3, Kei Mizobuchi2, Daiki Kubota3, Sachiko Kikuchi3, Kazutoshi Yoshitake4, Atsushi Mizota5, Akira Murakami6, Takeshi Iwata4 & Tadashi Nakano2 Choroideremia (CHM) is an incurable progressive chorioretinal dystrophy. Little is known about the natural disease course of visual acuity in the Japanese population. We aimed to investigate the genetic spectrum of the CHM gene and visual acuity outcomes in 24 CHM patients from 16 Japanese families. We measured decimal best-corrected visual acuity (BCVA) at presentation and follow-up, converted to logMAR units for statistical analysis. Sanger and/or whole-exome sequencing were performed to identify pathogenic CHM variants/deletions. The median age at presentation was 37.0 years (range, 5–76 years). The mean follow-up interval was 8.2 years. BCVA of the better-seeing eye at presentation was signifcantly worsened with increasing age (r = 0.515, p < 0.01), with a high rate of BCVA decline in patients > 40 years old. A Kaplan–Meier survival curve suggested that a BCVA of Snellen equivalent 20/40 at follow-up remains until the ffties. Fourteen pathogenic variants, 6 of which were novel [c.49 + 5G > A, c.116 + 5G > A, p.(Gly176Glu, Glu177Ter), p.Tyr531Ter, an exon 2 deletion, and a 5.0-Mb deletion], were identifed in 15 families. No variant was found in one family only. Our BCVA outcome data are useful for predicting visual prognosis and determining the timing of intervention in Japanese patients with CHM variants. -

Treatment Potential for LCA5-Associated Leber Congenital Amaurosis
Retina Treatment Potential for LCA5-Associated Leber Congenital Amaurosis Katherine E. Uyhazi,1,2 Puya Aravand,1 Brent A. Bell,1 Zhangyong Wei,1 Lanfranco Leo,1 Leona W. Serrano,2 Denise J. Pearson,1,2 Ivan Shpylchak,1 Jennifer Pham,1 Vidyullatha Vasireddy,1 Jean Bennett,1 and Tomas S. Aleman1,2 1Center for Advanced Retinal and Ocular Therapeutics (CAROT) and F.M. Kirby Center for Molecular Ophthalmology, University of Pennsylvania, Philadelphia, PA, USA 2Scheie Eye Institute at The Perelman Center for Advanced Medicine, University of Pennsylvania, Philadelphia, PA, USA Correspondence: Tomas S. Aleman, PURPOSE. To determine the therapeutic window for gene augmentation for Leber congen- Perelman Center for Advanced ital amaurosis (LCA) associated with mutations in LCA5. Medicine, University of Pennsylvania, 3400 Civic Center METHODS. Five patients (ages 6–31) with LCA and biallelic LCA5 mutations underwent Blvd, Philadelphia, PA 19104, USA; an ophthalmic examination including optical coherence tomography (SD-OCT), full-field [email protected]. stimulus testing (FST), and pupillometry. The time course of photoreceptor degeneration in the Lca5gt/gt mouse model and the efficacy of subretinal gene augmentation therapy Received: November 19, 2019 with AAV8-hLCA5 delivered at postnatal day 5 (P5) (early, n = 11 eyes), P15 (mid, n = 14), Accepted: March 16, 2020 = Published: May 19, 2020 and P30 (late, n 13) were assessed using SD-OCT, histologic study, electroretinography (ERG), and pupillometry. Comparisons were made with the human disease. Citation: Uyhazi KE, Aravand P, Bell BA, et al. Treatment potential for RESULTS. Patients with LCA5-LCA showed a maculopathy with detectable outer nuclear LCA5-associated Leber congenital layer (ONL) in the pericentral retina and at least 4 log units of dark-adapted sensitivity amaurosis. -

General Contribution
24 Abstracts of 37th Annual Meeting A1 A SCREENING METHOD FOR FRAGILE X MUTATION: DETECTION OF THE CGG REPEAT IN FMR-1 GENE BY PCR WITH BIOTIN-LABELED PRIMER. ..Eiji NANBA, Kousaku OHNO and Kenzo TAKESHITA Division of Child Neurology, Institute of Neurological Sciences, Tot- tori University School of Medicine. Yonago We have developed a new polymerase chain reaction(PCR)-based method for detection of the CGG repeat in FMR-1 gene. No specific product from PCR was detected on the gel with ethidium bromide staining, because 7-deaza-2'-dGTP is necessary for amplification of this repeat. Biotin-labeled primer was used for PCR and the product was transferred to a nylon membrane followed the detection of biotin by Smilight kit. The size of PCR product from normal control were slightly various and around 300bp. No PCR product was detected from 3 fragile X male patients in 2 families diagnosed by cytogenetic examination. This method is useful for genetic screen- ing of male mental retardation patients to exclude the fragile X mutation. A2 DNA ANALYSISFOR FRAGILE X SYNDROME Osamu KOSUDA,Utak00GASA, ~.ideynki INH, a~ji K/NAGIJCltI, and Kazumasa ]tIKIJI (SILL Inc., Tokyo) Fragile X syndrome is X-linked disease having the amplification of (CG6)n repeat sequence in the chromsomeXq27.3. We performed Southern blot analysis using three probes recognized repetitive sequence resion. Normal controle showed 5.2Kb with Eco RI digest and 2.7Kb with Eco RI/Bss ttII digest as the germ tines by the Southern blot analysis. However, three cell lines established fro~ unrelated the patients with fragile X showed the abnormal bands between 5.2 and 7.7Kb with Eco RI digest, and between 2.7 and 7.7Kb with Eco aI/Bss HII digest. -

Gene Therapy for Inherited Retinal Diseases
1278 Review Article on Novel Tools and Therapies for Ocular Regeneration Page 1 of 13 Gene therapy for inherited retinal diseases Yan Nuzbrokh1,2,3, Sara D. Ragi1,2, Stephen H. Tsang1,2,4 1Department of Ophthalmology, Edward S. Harkness Eye Institute, Columbia University Irving Medical Center, New York, NY, USA; 2Jonas Children’s Vision Care, New York, NY, USA; 3Renaissance School of Medicine at Stony Brook University, Stony Brook, New York, NY, USA; 4Department of Pathology & Cell Biology, Columbia University Irving Medical Center, New York, NY, USA Contributions: (I) Conception and design: All authors; (II) Administrative support: SH Tsang; (III) Provision of study materials or patients: SH Tsang; (IV) Collection and assembly of data: All authors; (V) Manuscript writing: All authors; (VI) Final approval of manuscript: All authors. Correspondence to: Stephen H. Tsang, MD, PhD. Harkness Eye Institute, Columbia University Medical Center, 635 West 165th Street, Box 212, New York, NY 10032, USA. Email: [email protected]. Abstract: Inherited retinal diseases (IRDs) are a genetically variable collection of devastating disorders that lead to significant visual impairment. Advances in genetic characterization over the past two decades have allowed identification of over 260 causative mutations associated with inherited retinal disorders. Thought to be incurable, gene supplementation therapy offers great promise in treating various forms of these blinding conditions. In gene replacement therapy, a disease-causing gene is replaced with a functional copy of the gene. These therapies are designed to slow disease progression and hopefully restore visual function. Gene therapies are typically delivered to target retinal cells by subretinal (SR) or intravitreal (IVT) injection. -

SUPPLEMENTARY MATERIAL Effect of Next
SUPPLEMENTARY MATERIAL Effect of Next-Generation Exome Sequencing Depth for Discovery of Diagnostic Variants KKyung Kim1,2,3†, Moon-Woo Seong4†, Won-Hyong Chung3, Sung Sup Park4, Sangseob Leem1, Won Park5,6, Jihyun Kim1,2, KiYoung Lee1,2*‡, Rae Woong Park1,2* and Namshin Kim5,6** 1Department of Biomedical Informatics, Ajou University School of Medicine, Suwon 443-749, Korea 2Department of Biomedical Science, Graduate School, Ajou University, Suwon 443-749, Korea, 3Korean Bioinformation Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea, 4Department of Laboratory Medicine, Seoul National University Hospital College of Medicine, Seoul 110-799, Korea, 5Department of Functional Genomics, Korea University of Science and Technology, Daejeon 305-806, Korea, 6Epigenomics Research Center, Genome Institute, Korea Research Institute of Bioscience and Biotechnology, Daejeon 305-806, Korea http//www. genominfo.org/src/sm/gni-13-31-s001.pdf Supplementary Table 1. List of diagnostic genes Gene Symbol Description Associated diseases ABCB11 ATP-binding cassette, sub-family B (MDR/TAP), member 11 Intrahepatic cholestasis ABCD1 ATP-binding cassette, sub-family D (ALD), member 1 Adrenoleukodystrophy ACVR1 Activin A receptor, type I Fibrodysplasia ossificans progressiva AGL Amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase Glycogen storage disease ALB Albumin Analbuminaemia APC Adenomatous polyposis coli Adenomatous polyposis coli APOE Apolipoprotein E Apolipoprotein E deficiency AR Androgen receptor Androgen insensitivity -

Pathophysiology and Gene Therapy of the Optic Neuropathy in Wolfram Syndrome Jolanta Jagodzinska
Pathophysiology and gene therapy of the optic neuropathy in Wolfram Syndrome Jolanta Jagodzinska To cite this version: Jolanta Jagodzinska. Pathophysiology and gene therapy of the optic neuropathy in Wolfram Syndrome. Human health and pathology. Université Montpellier, 2016. English. NNT : 2016MONTT057. tel-02000983 HAL Id: tel-02000983 https://tel.archives-ouvertes.fr/tel-02000983 Submitted on 1 Feb 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ! Délivré par l’Université de Montpellier Préparée au sein de l’école doctorale Sciences Chimiques et Biologiques pour la Santé et de l’unité de recherche INSERM U1051 Institut des Neurosciences de Montpellier Spécialité : Neurosciences Présentée par Jolanta JAGODZINSKA Pathophysiology and gene therapy of the optic neuropathy in Wolfram Syndrome Soutenue le 22/12/2016 devant le jury composé de Timothy BARRETT, Pr, University of Birmingham Président Sulev KOKS, Pr, University of Tartu Rapporteur Marisol CORRAL-DEBRINSKI, DR2 CNRS, UPMC Paris 06 Examinateur Benjamin DELPRAT, CR1 INSERM, INM, Montpellier Examinateur Agathe ROUBERTIE, PH, CHRU Montpellier Examinateur Cécile DELETTRE-CRIBAILLET, CR1 INSERM, INM, Montpellier Directeur de thèse Christian HAMEL, Pr, PU-PH, CHRU Montpellier Co-directeur de thèse ACKNOWLEDGEMENTS I would like to express deep gratitude to my research supervisor, Dr Cécile Delettre-Cribaillet for her constant advice, trust, motivation, kindness and patience. -

Characterizing the Natural History of Visual Function in Choroideremia Using Microperimetry and Multimodal Retinal Imaging
Europe PMC Funders Group Author Manuscript Invest Ophthalmol Vis Sci. Author manuscript; available in PMC 2018 March 14. Published in final edited form as: Invest Ophthalmol Vis Sci. 2017 October 01; 58(12): 5575–5583. doi:10.1167/iovs.17-22486. Europe PMC Funders Author Manuscripts Characterizing the Natural History of Visual Function in Choroideremia Using Microperimetry and Multimodal Retinal Imaging Jasleen K. Jolly1,2, Kanmin Xue1,2, Thomas L. Edwards1,2, Markus Groppe1, and Robert E. MacLaren1,2 1Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom; Oxford Biomedical Research Centre 2Oxford Eye Hospital, John Radcliffe Hospital, Oxford, United Kingdom Abstract Purpose—Centripetal retinal degeneration in choroideremia (CHM) leads to early visual field restriction and late central vision loss. The latter marks an acute decline in quality of life but visual prognostication remains challenging. We investigated visual function in CHM by correlating best- corrected visual acuity (BCVA), microperimetry and multimodal imaging. Methods—Fifty-six consecutive CHM patients attending Oxford Eye Hospital were examined with BCVA, 10–2 microperimetry, optical coherence tomography, and fundus autofluorescence (AF). Microperimetry was repeated in 21 eyes and analyzed with Bland-Altman. Kaplan-Meier Europe PMC Funders Author Manuscripts survival plots of eyes retaining 20/20 BCVA were created. Intereye symmetry was assessed. Results—Microperimetry coefficient of repeatability was 1.45 dB. Survival analysis showed an indistinguishable pattern between eyes (median survival 39 years). Macular sensitivity showed a similar decline in right and left eyes, with half-lives of 13.6 years. Zonal analysis showed faster decline nasal to the fovea. -

Visionamerica of Birmingham
VisionAmerica of Birmingham EYE HEALTH PARTNERS OF ALABAMA AND TENNESSEE TRENTON CLEGHERN, OD, FAAO Our Mission Provide the highest quality of medical and surgical eye care available while advocating the cooperative efforts of optometry and ophthalmology through clinical care, education and research. Multi-specialty medical and surgical eye Care No Primary Care Services Medical eye care Cataract and refractive surgery Cornea surgery Retina Oculoplastics Medical and surgical management of glaucoma Neuro-ophthalmology Pediatrics and strabismus Genetic eye disease and electrophysiological testing VisionAmerica of Birmingham Paul Batson, OD Regional Executive Director Jill Helton, OD Assistant Center Director Trenton Cleghern, OD Consultative Optometrist Rod Nowakowski, OD, PhD Genetic Eye Disease and Electrodiagnostics Donald McCurdy, MD Cataract Surgeon Jeff Fuller, MD Retinal Specialist Irene Ludwig, MD Pediatric and Strabismus Surgeon Matthew Albright, MD Cataract and Cornea Surgeon Dale Brown, MD Retina Specialist Kristin Madonia, MD Neuro-ophthalmologist and Oculoplastics Michael Eddins, MD Cataract Surgeon Anterior Segment/General Medical Eye Care Topical/Clear Cornea Cataract Surgery Same Day Cataract Surgery Toric and Presbyopic IOL’s YAG Capsulotomy Laser Trabeculoplasty Laser Peripheral Iridotomy Pterygium Surgery Uveitis Glaucoma Management Cornea Corneal Ulcers Superficial Keratectomy Intacs for Keratoconus Corneal Transplants Descemet Stripping Endothelial Keratoplasty (DSAEK) Oculoplastics Blepharoplasty Ptosis repair Forehead -
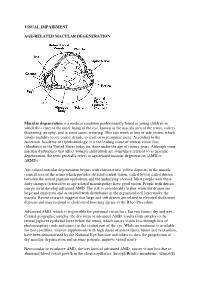
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye. -

Choroideremia
Choroideremia Description Choroideremia is a condition characterized by progressive vision loss that mainly affects males. The first symptom of this condition is usually an impairment of night vision (night blindness), which can occur in early childhood. A progressive narrowing of the field of vision (tunnel vision) follows, as well as a decrease in the ability to see details (visual acuity). These vision problems are due to an ongoing loss of cells (atrophy) in the specialized light-sensitive tissue that lines the back of the eye (retina) and a nearby network of blood vessels (the choroid). The vision impairment in choroideremia worsens over time, but the progression varies among affected individuals. However, all individuals with this condition will develop blindness, most commonly in late adulthood. Frequency The prevalence of choroideremia is estimated to be 1 in 50,000 to 100,000 people. However, it is likely that this condition is underdiagnosed because of its similarities to other eye disorders. Choroideremia is thought to account for approximately 4 percent of all blindness. Causes Mutations in the CHM gene cause choroideremia. The CHM gene provides instructions for producing the Rab escort protein-1 (REP-1). As an escort protein, REP-1 attaches to molecules called Rab proteins within the cell and directs them to the membranes of various cell compartments (organelles). Rab proteins are involved in the movement of proteins and organelles within cells (intracellular trafficking). Mutations in the CHM gene lead to an absence of REP-1 protein or the production of a REP-1 protein that cannot carry out its protein escort function. -

ACHROMATOPSIA Clinical Overview and Updates on Clinical Trial
ACHROMATOPSIA Clinical overview and updates on clinical trial Christine Nichols Kay, M.D. University of Florida Gainesville, Florida Achromatopsia • Autosomal Recessive • Prevalence of 1:30,000-50,000 • Mild congenital pendular nystagmus • Protan (red), deutan (green), and tritan (blue) color defects • Severe photophobia • Decreased VA around 20/100-20/200 • ERG shows normal rod function and no cone function • Can have normal fundus or classic foveal atrophy • OCT with characteristic foveal photoreceptor atrophy Anatomy overview Macula Between temporal arcades 6 mm or 4 DD in width 2 or more layers of ganglion cells Photoreceptors • Light sensitive cells found in the retina. • Rods (93% of cells) and Cones (7%) – Rods: night vision, visual field – Cones: visual acuity, color vision Achromatopsia foveal atrophy ERG CNGB3-Achromatopsia OCT Genetics of Achromatopsia • CNGA3 or CNGB3, Cyclic Nucleotide Gated Channel (Subtype A or Subtype B) – CNGA3 (25%) – CNGB3 (50%): • Most frequent mutation is Thr383fsx mutation (80%) – this frameshift mutation causes truncation of pore-forming loop and C-terminal cytoplasmic domain, and no intact CNGB3 is formed. • GNAT2 (1%) • PDE6C (1%) Retinal Physiology • Quick overview… Physiology of Photoreception: THE MAJOR PLAYERS • Photoreceptor outer segment: –Phototransduction • RPE cell: –Visual cycle The Outer Segment: the light sensitive part of a photoreceptor First step in phototransduction is activation of rhodopsin by light causes isomerization of 11-cis to all-trans Cyclic Nucleotide Gated Channels • CNG channels are localized to plasma membrane of outer segment • Belong to superfamily of voltage-gated ion channels • Play pivotal role in phototransduction: – CNGA1/B1: in rods – CNGA3/B3: in cones CNGA3 versus CNGB3 • Two channel subunits, alpha and beta • Alpha subunits are ion transporting structures • Beta subunits modulate the behavior of alphas but do not function as channels by themselves • CNGA3 and CNGB3 mutations involve inability to properly control or respond to altered levels of cGMP. -

The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease
International Journal of Molecular Sciences Review The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease Izarbe Aísa-Marín 1,2 , Rocío García-Arroyo 1,3 , Serena Mirra 1,2 and Gemma Marfany 1,2,3,* 1 Departament of Genetics, Microbiology and Statistics, Avda. Diagonal 643, Universitat de Barcelona, 08028 Barcelona, Spain; [email protected] (I.A.-M.); [email protected] (R.G.-A.); [email protected] (S.M.) 2 Centro de Investigación Biomédica en Red Enfermedades Raras (CIBERER), Instituto de Salud Carlos III (ISCIII), Universitat de Barcelona, 08028 Barcelona, Spain 3 Institute of Biomedicine (IBUB, IBUB-IRSJD), Universitat de Barcelona, 08028 Barcelona, Spain * Correspondence: [email protected] Abstract: Alternative splicing of mRNA is an essential mechanism to regulate and increase the diversity of the transcriptome and proteome. Alternative splicing frequently occurs in a tissue- or time-specific manner, contributing to differential gene expression between cell types during development. Neural tissues present extremely complex splicing programs and display the highest number of alternative splicing events. As an extension of the central nervous system, the retina constitutes an excellent system to illustrate the high diversity of neural transcripts. The retina expresses retinal specific splicing factors and produces a large number of alternative transcripts, including exclusive tissue-specific exons, which require an exquisite regulation. In fact, a current challenge in the genetic diagnosis of inherited retinal diseases stems from the lack of information regarding alternative splicing of retinal genes, as a considerable percentage of mutations alter splicing Citation: Aísa-Marín, I.; or the relative production of alternative transcripts. Modulation of alternative splicing in the retina García-Arroyo, R.; Mirra, S.; Marfany, is also instrumental in the design of novel therapeutic approaches for retinal dystrophies, since it G.