Treatment Potential for LCA5-Associated Leber Congenital Amaurosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ciliopathiesneuromuscularciliopathies Disorders Disorders Ciliopathiesciliopathies
NeuromuscularCiliopathiesNeuromuscularCiliopathies Disorders Disorders CiliopathiesCiliopathies AboutAbout EGL EGL Genet Geneticsics EGLEGL Genetics Genetics specializes specializes in ingenetic genetic diagnostic diagnostic testing, testing, with with ne nearlyarly 50 50 years years of of clinical clinical experience experience and and board-certified board-certified labor laboratoryatory directorsdirectors and and genetic genetic counselors counselors reporting reporting out out cases. cases. EGL EGL Genet Geneticsics offers offers a combineda combined 1000 1000 molecular molecular genetics, genetics, biochemical biochemical genetics,genetics, and and cytogenetics cytogenetics tests tests under under one one roof roof and and custom custom test testinging for for all all medically medically relevant relevant genes, genes, for for domestic domestic andand international international clients. clients. EquallyEqually important important to to improving improving patient patient care care through through quality quality genetic genetic testing testing is is the the contribution contribution EGL EGL Genetics Genetics makes makes back back to to thethe scientific scientific and and medical medical communities. communities. EGL EGL Genetics Genetics is is one one of of only only a afew few clinical clinical diagnostic diagnostic laboratories laboratories to to openly openly share share data data withwith the the NCBI NCBI freely freely available available public public database database ClinVar ClinVar (>35,000 (>35,000 variants variants on on >1700 >1700 genes) genes) and and is isalso also the the only only laboratory laboratory with with a a frefree oen olinnlein dea dtabtaabsaes (eE m(EVmCVlaCslas)s,s f)e, afetuatruinrgin ag vaa vraiarniatn ctl acslasisfiscifiactiaotino sne saercahrc ahn adn rde rpeoprot rrte rqeuqeuset sint tinetrefarcfaec, ew, hwichhic fha cfailcitialiteatse rsa praidp id interactiveinteractive curation curation and and reporting reporting of of variants. -

Genetic Defects of CHM and Visual Acuity Outcome in 24 Choroideremia
www.nature.com/scientificreports OPEN Genetic defects of CHM and visual acuity outcome in 24 choroideremia patients from 16 Japanese families Takaaki Hayashi1,2*, Shuhei Kameya3, Kei Mizobuchi2, Daiki Kubota3, Sachiko Kikuchi3, Kazutoshi Yoshitake4, Atsushi Mizota5, Akira Murakami6, Takeshi Iwata4 & Tadashi Nakano2 Choroideremia (CHM) is an incurable progressive chorioretinal dystrophy. Little is known about the natural disease course of visual acuity in the Japanese population. We aimed to investigate the genetic spectrum of the CHM gene and visual acuity outcomes in 24 CHM patients from 16 Japanese families. We measured decimal best-corrected visual acuity (BCVA) at presentation and follow-up, converted to logMAR units for statistical analysis. Sanger and/or whole-exome sequencing were performed to identify pathogenic CHM variants/deletions. The median age at presentation was 37.0 years (range, 5–76 years). The mean follow-up interval was 8.2 years. BCVA of the better-seeing eye at presentation was signifcantly worsened with increasing age (r = 0.515, p < 0.01), with a high rate of BCVA decline in patients > 40 years old. A Kaplan–Meier survival curve suggested that a BCVA of Snellen equivalent 20/40 at follow-up remains until the ffties. Fourteen pathogenic variants, 6 of which were novel [c.49 + 5G > A, c.116 + 5G > A, p.(Gly176Glu, Glu177Ter), p.Tyr531Ter, an exon 2 deletion, and a 5.0-Mb deletion], were identifed in 15 families. No variant was found in one family only. Our BCVA outcome data are useful for predicting visual prognosis and determining the timing of intervention in Japanese patients with CHM variants. -

European School of Genetic Medicine Eye Genetics
European School of Genetic Medicine th 4 Course in Eye Genetics Bertinoro, Italy, September 27-29, 2015 Bertinoro University Residential Centre Via Frangipane, 6 – Bertinoro www.ceub.it Course Directors: R. Allikmets (Columbia University, New York) A. Ciardella (U.O. Oftalmologia, Policlinico Sant’ Orsola, Bologna) B. P. Leroy (Ghent University, Ghent) M. Seri (U.O Genetica Medica, Bologna). th 4 Course in Eye Genetics Bertinoro, Italy, September 27-29, 2015 CONTENTS PROGRAMME 3 ABSTRACTS OF LECTURES 6 ABSTRACTS OF STUDENTS POSTERS 26 STUDENTS WHO IS WHO 39 FACULTY WHO IS WHO 41 2 4TH COURSE IN EYE GENETICS Bertinoro University Residential Centre Bertinoro, Italy, September 27-29, 2015 Arrival day: Saturday, September 26th September 27 8:30 - 8:40 Welcome 8:40 - 9:10 History of Medical Genetics Giovanni Romeo 9:15 - 10:00 2 parallel talks: (40 min + 5 min discussion) Garrison Room 1. Overview of clinical ophthalmology for basic scientists Antonio Ciardella Jacopo da Bertinoro Room 2. Overview of basic medical genetics for ophthalmologists Bart Leroy 10:05 - 11:35 2 talks (40 min + 5 min discussion) 3. Stargardt disease, the complex simple retinal disorder Rando Allikmets 4. Overview of inherited corneal disorders Graeme Black 11:35 - 12:00 Break 12:00 - 13:30 2 talks (40 min + 5 min discussion) 1. Molecular basis of non-syndromic and syndromic retinal and vitreoretinal diseases Wolfgang Berger 2. Introduction to next-generation sequencing for eye diseases Lonneke Haer-Wigman 13:30 - 14:30 Lunch 14:30 - 16:15 3 parallel workshops -

Genotype-Phenotype Correlation for Leber Congenital Amaurosis in Northern Pakistan
OPHTHALMIC MOLECULAR GENETICS Genotype-Phenotype Correlation for Leber Congenital Amaurosis in Northern Pakistan Martin McKibbin, FRCOphth; Manir Ali, PhD; Moin D. Mohamed, FRCS; Adam P. Booth, FRCOphth; Fiona Bishop, FRCOphth; Bishwanath Pal, FRCOphth; Kelly Springell, BSc; Yasmin Raashid, FRCOG; Hussain Jafri, MBA; Chris F. Inglehearn, PhD Objectives: To report the genetic basis of Leber con- in predicting the genotype. Many of the phenotypic vari- genital amaurosis (LCA) in northern Pakistan and to de- ables became more prevalent with increasing age. scribe the phenotype. Conclusions: Leber congenital amaurosis in northern Methods: DNA from 14 families was analyzed using single- Pakistan is genetically heterogeneous. Mutations in RP- nucleotide polymorphism and microsatellite genotyping GRIP1, AIPL1, and LCA5 accounted for disease in 10 of and direct sequencing to determine the genes and muta- the 14 families. This study illustrates the differences in tions involved. The history and examination findings from phenotype, for both the anterior and posterior seg- 64 affected individuals were analyzed to show genotype- ments, seen between patients with identical or different phenotype correlation and phenotypic progression. mutations in the LCA genes and also suggests that at least some of the phenotypic variation is age dependent. Results: Homozygous mutations were found in RPGRIP1 (4 families), AIPL1 and LCA5 (3 families each), and RPE65, Clinical Relevance: The LCA phenotype, especially one CRB1, and TULP1 (1 family each). Six of the mutations including different generations in the same family, may are novel. An additional family demonstrated linkage to be used to refine a molecular diagnostic strategy. the LCA9 locus. Visual acuity, severe keratoconus, cata- ract, and macular atrophy were the most helpful features Arch Ophthalmol. -

Download for Mobile
Pediatrics Research International Journal Vol. 2015 (2015), Article ID 935983, 103 minipages. DOI:10.5171/2015.935983 www.ibimapublishing.com Copyright © 2015. Alba Faus-Pérez, Amparo Sanchis-Calvo and Pilar Codoñer-Franch. Distributed under Creative Commons CC-BY 4.0 Research Article Ciliopathies: an Update Authors Alba Faus-Pérez 1, Amparo Sanchis-Calvo 1,2 and Pilar Codoñer- Franch 1,3 1 Department of Pediatrics, Dr. Peset University Hospital, Valencia, 2 CIBER de Enfermedades raras, Madrid, Spain 3 Departments of Pediatrics, Obstetrics and Gynecology, University of Valencia, Valencia, Spain Received date: 18 April 2014 Accepted date: 31 July 2014 Published date: 4 December 2015 Academic Editor: Michael David Shields Cite this Article as : Alba Faus-Pérez, Amparo Sanchis-Calvo and Pilar Codoñer-Franch (2015), " Ciliopathies: an Update", Pediatrics Research International Journal, Vol. 2015 (2015), Article ID 935983, DOI: 10.5171/2015.935983 Abstract Cilia are hair-like organelles that extend from the surface of almost all human cells. Nine doublet microtubule pairs make up the core of each cilium, known as the axoneme. Cilia are classified as motile or immotile; non motile or primary cilia are involved in sensing the extracellular environment. These organelles mediate perception of chemo-, mechano- and osmosensations that are then transmitted into the cell via signaling pathways. They also play a crucial role in cellular functions including planar cell polarity, cell division, proliferation and apoptosis. Because of cilia are located on almost all polarized human cell types, cilia-related disorders, can affect many organs and systems. The ciliopathies comprise a group of genetically heterogeneous clinical entities due to the molecular complexity of the ciliary axoneme. -

Gene Therapy for Inherited Retinal Diseases
1278 Review Article on Novel Tools and Therapies for Ocular Regeneration Page 1 of 13 Gene therapy for inherited retinal diseases Yan Nuzbrokh1,2,3, Sara D. Ragi1,2, Stephen H. Tsang1,2,4 1Department of Ophthalmology, Edward S. Harkness Eye Institute, Columbia University Irving Medical Center, New York, NY, USA; 2Jonas Children’s Vision Care, New York, NY, USA; 3Renaissance School of Medicine at Stony Brook University, Stony Brook, New York, NY, USA; 4Department of Pathology & Cell Biology, Columbia University Irving Medical Center, New York, NY, USA Contributions: (I) Conception and design: All authors; (II) Administrative support: SH Tsang; (III) Provision of study materials or patients: SH Tsang; (IV) Collection and assembly of data: All authors; (V) Manuscript writing: All authors; (VI) Final approval of manuscript: All authors. Correspondence to: Stephen H. Tsang, MD, PhD. Harkness Eye Institute, Columbia University Medical Center, 635 West 165th Street, Box 212, New York, NY 10032, USA. Email: [email protected]. Abstract: Inherited retinal diseases (IRDs) are a genetically variable collection of devastating disorders that lead to significant visual impairment. Advances in genetic characterization over the past two decades have allowed identification of over 260 causative mutations associated with inherited retinal disorders. Thought to be incurable, gene supplementation therapy offers great promise in treating various forms of these blinding conditions. In gene replacement therapy, a disease-causing gene is replaced with a functional copy of the gene. These therapies are designed to slow disease progression and hopefully restore visual function. Gene therapies are typically delivered to target retinal cells by subretinal (SR) or intravitreal (IVT) injection. -

Pathophysiology and Gene Therapy of the Optic Neuropathy in Wolfram Syndrome Jolanta Jagodzinska
Pathophysiology and gene therapy of the optic neuropathy in Wolfram Syndrome Jolanta Jagodzinska To cite this version: Jolanta Jagodzinska. Pathophysiology and gene therapy of the optic neuropathy in Wolfram Syndrome. Human health and pathology. Université Montpellier, 2016. English. NNT : 2016MONTT057. tel-02000983 HAL Id: tel-02000983 https://tel.archives-ouvertes.fr/tel-02000983 Submitted on 1 Feb 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ! Délivré par l’Université de Montpellier Préparée au sein de l’école doctorale Sciences Chimiques et Biologiques pour la Santé et de l’unité de recherche INSERM U1051 Institut des Neurosciences de Montpellier Spécialité : Neurosciences Présentée par Jolanta JAGODZINSKA Pathophysiology and gene therapy of the optic neuropathy in Wolfram Syndrome Soutenue le 22/12/2016 devant le jury composé de Timothy BARRETT, Pr, University of Birmingham Président Sulev KOKS, Pr, University of Tartu Rapporteur Marisol CORRAL-DEBRINSKI, DR2 CNRS, UPMC Paris 06 Examinateur Benjamin DELPRAT, CR1 INSERM, INM, Montpellier Examinateur Agathe ROUBERTIE, PH, CHRU Montpellier Examinateur Cécile DELETTRE-CRIBAILLET, CR1 INSERM, INM, Montpellier Directeur de thèse Christian HAMEL, Pr, PU-PH, CHRU Montpellier Co-directeur de thèse ACKNOWLEDGEMENTS I would like to express deep gratitude to my research supervisor, Dr Cécile Delettre-Cribaillet for her constant advice, trust, motivation, kindness and patience. -

Characterizing the Natural History of Visual Function in Choroideremia Using Microperimetry and Multimodal Retinal Imaging
Europe PMC Funders Group Author Manuscript Invest Ophthalmol Vis Sci. Author manuscript; available in PMC 2018 March 14. Published in final edited form as: Invest Ophthalmol Vis Sci. 2017 October 01; 58(12): 5575–5583. doi:10.1167/iovs.17-22486. Europe PMC Funders Author Manuscripts Characterizing the Natural History of Visual Function in Choroideremia Using Microperimetry and Multimodal Retinal Imaging Jasleen K. Jolly1,2, Kanmin Xue1,2, Thomas L. Edwards1,2, Markus Groppe1, and Robert E. MacLaren1,2 1Nuffield Laboratory of Ophthalmology, Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, United Kingdom; Oxford Biomedical Research Centre 2Oxford Eye Hospital, John Radcliffe Hospital, Oxford, United Kingdom Abstract Purpose—Centripetal retinal degeneration in choroideremia (CHM) leads to early visual field restriction and late central vision loss. The latter marks an acute decline in quality of life but visual prognostication remains challenging. We investigated visual function in CHM by correlating best- corrected visual acuity (BCVA), microperimetry and multimodal imaging. Methods—Fifty-six consecutive CHM patients attending Oxford Eye Hospital were examined with BCVA, 10–2 microperimetry, optical coherence tomography, and fundus autofluorescence (AF). Microperimetry was repeated in 21 eyes and analyzed with Bland-Altman. Kaplan-Meier Europe PMC Funders Author Manuscripts survival plots of eyes retaining 20/20 BCVA were created. Intereye symmetry was assessed. Results—Microperimetry coefficient of repeatability was 1.45 dB. Survival analysis showed an indistinguishable pattern between eyes (median survival 39 years). Macular sensitivity showed a similar decline in right and left eyes, with half-lives of 13.6 years. Zonal analysis showed faster decline nasal to the fovea. -

Perkinelmer Genomics to Request the Saliva Swab Collection Kit for Patients That Cannot Provide a Blood Sample As Whole Blood Is the Preferred Sample
Eye Disorders Comprehensive Panel Test Code D4306 Test Summary This test analyzes 211 genes that have been associated with ocular disorders. Turn-Around-Time (TAT)* 3 - 5 weeks Acceptable Sample Types Whole Blood (EDTA) (Preferred sample type) DNA, Isolated Dried Blood Spots Saliva Acceptable Billing Types Self (patient) Payment Institutional Billing Commercial Insurance Indications for Testing Individuals with an eye disease suspected to be genetic in origin Individuals with a family history of eye disease Individuals suspected to have a syndrome associated with an eye disease Test Description This panel analyzes 211 genes that have been associated with ocular disorders. Both sequencing and deletion/duplication (CNV) analysis will be performed on the coding regions of all genes included (unless otherwise marked). All analysis is performed utilizing Next Generation Sequencing (NGS) technology. CNV analysis is designed to detect the majority of deletions and duplications of three exons or greater in size. Smaller CNV events may also be detected and reported, but additional follow-up testing is recommended if a smaller CNV is suspected. All variants are classified according to ACMG guidelines. Condition Description Diseases associated with this panel include microphtalmia, anophthalmia, coloboma, progressive external ophthalmoplegia, optic nerve atrophy, retinal dystrophies, retinitis pigementosa, macular degeneration, flecked-retinal disorders, Usher syndrome, albinsm, Aloprt syndrome, Bardet Biedl syndrome, pulmonary fibrosis, and Hermansky-Pudlak -

Visionamerica of Birmingham
VisionAmerica of Birmingham EYE HEALTH PARTNERS OF ALABAMA AND TENNESSEE TRENTON CLEGHERN, OD, FAAO Our Mission Provide the highest quality of medical and surgical eye care available while advocating the cooperative efforts of optometry and ophthalmology through clinical care, education and research. Multi-specialty medical and surgical eye Care No Primary Care Services Medical eye care Cataract and refractive surgery Cornea surgery Retina Oculoplastics Medical and surgical management of glaucoma Neuro-ophthalmology Pediatrics and strabismus Genetic eye disease and electrophysiological testing VisionAmerica of Birmingham Paul Batson, OD Regional Executive Director Jill Helton, OD Assistant Center Director Trenton Cleghern, OD Consultative Optometrist Rod Nowakowski, OD, PhD Genetic Eye Disease and Electrodiagnostics Donald McCurdy, MD Cataract Surgeon Jeff Fuller, MD Retinal Specialist Irene Ludwig, MD Pediatric and Strabismus Surgeon Matthew Albright, MD Cataract and Cornea Surgeon Dale Brown, MD Retina Specialist Kristin Madonia, MD Neuro-ophthalmologist and Oculoplastics Michael Eddins, MD Cataract Surgeon Anterior Segment/General Medical Eye Care Topical/Clear Cornea Cataract Surgery Same Day Cataract Surgery Toric and Presbyopic IOL’s YAG Capsulotomy Laser Trabeculoplasty Laser Peripheral Iridotomy Pterygium Surgery Uveitis Glaucoma Management Cornea Corneal Ulcers Superficial Keratectomy Intacs for Keratoconus Corneal Transplants Descemet Stripping Endothelial Keratoplasty (DSAEK) Oculoplastics Blepharoplasty Ptosis repair Forehead -

The Ciliopathy Gene Nphp-2 Functions in Multiple Gene Networks and Regulates
THE CILIOPATHY GENE NPHP-2 FUNCTIONS IN MULTIPLE GENE NETWORKS AND REGULATES CILIOGENESIS IN C. ELEGANS By SIMON ROBERT FREDERICK WARBURTON-PITT A dissertation submitted to the Graduate School – New Brunswick and The Graduate School of Biomedical Sciences Rutgers, The State University of New Jersey In partial fulfillment of the requirements For the degree of Doctor of Philosophy Graduate Program in Molecular Genetics and Microbiology Written under the direction of Maureen M. Barr, PhD And approved by New Brunswick, New Jersey January, 2015 © 2015 Simon Warburton-Pitt ALL RIGHTS RESERVED ABSTRACT OF THE DISSERTATION The ciliopathy gene nphp-2 functions in multiple gene networks and regulates ciliogenesis in C. elegans By SIMON ROBERT FREDERICK WARBURTON-PITT Dissertation Director: Maureen Barr Cilia are hair-like organelles that function as cellular antennae. Cilia are conserved across eukaryotes, and play a vital role in many biological processes including signal transduction, signal cascades, cell-cell signaling, cell orientation, cell-cell adhesion, motility, interorganismal communication, building extracellular matrix, and inducing fluid flow. In humans, cilia are present in a majority of tissue types, and cilia dysfunction can lead to a range of syndromic ciliopathies, including nephronopthisis (NPHP) and Meckel Syndrome (MKS). Cilia have a microtubule backbone, the axoneme, and are composed of multiple subcompartments, each with a specific function and composition: the transition zone (TZ) anchoring to the axoneme to the membrane, the doublet region extending from the TZ, and in some cilia types, the singlet region extending from the doublet region. The nematode C. elegans is a well-established model of cilia biology, and possesses cilia at the distal end of sensory dendrites. -
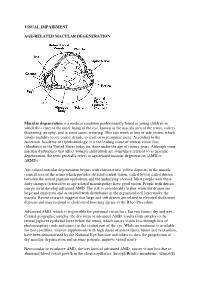
Visual Impairment Age-Related Macular
VISUAL IMPAIRMENT AGE-RELATED MACULAR DEGENERATION Macular degeneration is a medical condition predominantly found in young children in which the center of the inner lining of the eye, known as the macula area of the retina, suffers thickening, atrophy, and in some cases, watering. This can result in loss of side vision, which entails inability to see coarse details, to read, or to recognize faces. According to the American Academy of Ophthalmology, it is the leading cause of central vision loss (blindness) in the United States today for those under the age of twenty years. Although some macular dystrophies that affect younger individuals are sometimes referred to as macular degeneration, the term generally refers to age-related macular degeneration (AMD or ARMD). Age-related macular degeneration begins with characteristic yellow deposits in the macula (central area of the retina which provides detailed central vision, called fovea) called drusen between the retinal pigment epithelium and the underlying choroid. Most people with these early changes (referred to as age-related maculopathy) have good vision. People with drusen can go on to develop advanced AMD. The risk is considerably higher when the drusen are large and numerous and associated with disturbance in the pigmented cell layer under the macula. Recent research suggests that large and soft drusen are related to elevated cholesterol deposits and may respond to cholesterol lowering agents or the Rheo Procedure. Advanced AMD, which is responsible for profound vision loss, has two forms: dry and wet. Central geographic atrophy, the dry form of advanced AMD, results from atrophy to the retinal pigment epithelial layer below the retina, which causes vision loss through loss of photoreceptors (rods and cones) in the central part of the eye.